




















Puntos clave

Las enfermedades lisosomales (EL) se producen por un defecto genético que altera la síntesis o función de una enzima lisosomal específica o de su proteína transportadora. Se genera un depósito intralisosomal de moléculas complejas con anomalías orgánicas y funcionales progresivas y graves. Su presentación clínica es muy variable y su pronóstico, sin tratamiento específico, suele ser malo. Hasta el momento se han descrito unas 50 entidades diferentes1,2. De forma aislada son infrecuentes pero en conjunto su incidencia se estima en 1 por cada 5.000 o 7.000 recién nacidos3,4. Son enfermedades raras, infradiagnosticadas, que se manifiestan a cualquier edad, por lo que el pediatra debe estar familiarizado con ellas. El reciente desarrollo de nuevas terapias, como el tratamiento enzimático sustitutivo (TES) para algunas de ellas (Gaucher, Fabry, Pompe, mucopolisacaridosis [MPS] tipos I, II y VI), con demostrada eficacia y seguridad, subraya la necesidad del diagnóstico y tratamiento precoces de estos pacientes5.
Enfermedades lisosomalesLas EL se caracterizan por su afectación progresiva y multisistémica, dando lugar a fenotipos complejos con asociación de manifestaciones viscerales, esqueléticas y neurológicas. Se presentan a cualquier edad, desde la etapa prenatal hasta la época de adulto, y su expresividad es muy variable.
El punto de partida más útil para el diagnóstico de una EL es su sospecha clínica. La presencia anormal de glucosaminoglucanos (GAG), oligosacáridos o ácido siálico en orina sugieren la mayoría de MPS, mucolipidosis, glucoproteinosis, enfermedades por almacenamiento de ácido siálico, galactosialidosis y de deficiencia múltiple de sulfatasas. La confirmación diagnóstica se basa en la identificación del déficit enzimático en plasma, leucocitos o fibroblastos. El estudio genético identifica el defecto molecular subyacente, permitiendo el cribado de portadores en la familia y su asesoramiento genético adecuado. La mayoría de estas enfermedades se heredan de forma autosómica recesiva, excepto Fabry y MPS II que lo hacen de forma ligada a X. En los casos con riesgo significativo para la recurrencia de la enfermedad, se puede ofrecer diagnóstico prenatal enzimático y sólo cuando el defecto genético está identificado en la familia es posible el estudio molecular prenatal y/o preimplantacional.
Tratamiento en las enfermedades lisosomalesHasta hace poco sólo existía la posibilidad de tratamientos sintomáticos; sin embargo, en las últimas décadas se ha progresado mucho en el desarrollo de nuevas terapias como el trasplante de progenitores hematopoyéticos (TPH), el TES y la terapia de reducción de sustrato (TRS). Se sigue investigando en otras posibles modalidades terapéuticas como el uso de fármacos antiinflamatorios y terapia génica, entre otras.
Lectura rápida

Las enfermedades lisosomales (EL) se producen por un defecto genético que altera una enzima lisosomal o su proteína transportadora. Se genera un depósito intralisosomal de moléculas complejas con anomalías orgánicas y funcionales progresivas y graves.
Son enfermedades raras, hereditarias, la mayoría autosómicas recesivas. Se conocen más de 50 EL diferentes.
Enfermedades lisosomalesLa clínica de las EL es muy variable y su inicio es frecuente en la edad pediátrica. Se caracterizan por su afectación progresiva y multisistémica, dando lugar a fenotipos complejos con asociación de manifestaciones viscerales, esqueléticas y neurológicas, entre otras. El diagnóstico de sospecha es clínico y algunos exámenes complementarios pueden ayudar a su orientación sindrómica. La confirmación diagnóstica se basa en la identificación del déficit enzimático en plasma, leucocitos o fibroblastos. El estudio genético identifica el defecto molecular subyacente, permitiendo el cribado de portadores en la familia y su asesoramiento genético adecuado.

El TPH se usó como tratamiento de una EL por primera vez en 1980, utilizando médula ósea en un paciente con síndrome de Hurler6. Se basa en que las células del donante sustituyan a las células del paciente. Las células de la médula ósea pueden cruzar la barrera hematoencefálica (BHE), a diferencia del TES, y diferenciarse en células microgliales. Se ha empleado con éxito en la MPS tipo I, α-manosidosis, leucodistrofia metacromática y enfermedad de Krabbe, entre otras. Su realización precoz puede preservar la función neurológica. Sin embargo, el TPH presenta inconvenientes como su falta de efecto en algunas EL, en ciertas manifestaciones, en particular las esqueléticas, y su alta morbilidad y mortalidad (del 10-25%)5,7.
El TES consiste en la administración periódica por vía intravenosa, de forma hospitalaria o domiciliaria, de la enzima deficitaria, sintetizada por ingeniería genética y marcada con una señal bioquímica. De esta forma, la enzima exógena penetra en las células a través de los receptores manosa-6-fosfato (M6P) y alcanza los lisosomas. En las últimas décadas, se han desarrollado varias enzimas para TES. Su tolerancia es buena aunque, en ocasiones, pueden presentar prurito, fiebre o escalofríos, que suelen responder a la premedicación (antihistamínicos, corticoides y antitérmicos) y a la reducción de la velocidad de perfusión.
Las limitaciones del TES vienen determinadas por: a) las enzimas no atraviesan la BHE por su tamaño, por lo que no actúa en el sistema nervioso central; actualmente se están ensayando tratamientos intratecales; b) no penetra bien en los tejidos menos vascularizados como hueso, cartílago y válvulas cardíacas; c) origina anticuerpos que pueden influir en su eficacia, y d) su coste es muy alto1,2. Sin embargo, su efecto beneficioso y su tolerancia le hace hoy en día ser el tratamiento específico de elección en 6 enfermedades lisosomales que vamos a revisar a continuación: enfermedad de Gaucher, enfermedad de Fabry, MPS I, MPS II, MPS VI y enfermedad de Pompe (tabla 1).
Lectura rápida
La atención del paciente con EL debe ser multidisciplinaria, incluyendo un tratamiento sintomático y específico con el objetivo de mejorar su calidad de vida. Además se debe ofrecer asesoramiento genético a la familia.
El tratamiento enzimático sustitutivo (TES) ha demostrado su eficacia (mayor cuanto más precoz sea su instauración) y buena tolerancia en varias EL. Es el tratamiento específico de elección para: enfermedad de Gaucher, enfermedad de Fabry, mucopolisacaridosis (MPS) I, II y VI y enfermedad de Pompe.
Enfermedades lisosomales con tratamiento enzimático sustitutivoLa enfermedad de Gaucher es la más prevalente, se hereda de forma autosómica recesiva. El tipo 1 es el más frecuente. La afectación visceral, hematológica y ósea es característica. La afectación neurológica sólo se da en las formas graves de presentación precoz (tipos 2 y 3). El TES corrige las anomalías viscerales y hematológicas rápidamente, mejora o normaliza el crecimiento y revierte la afectación de la médula ósea, reduciendo la frecuencia de crisis óseas y de fracturas. En general, no se modifica la evolución neurológica.
Enfermedades lisosomales con tratamiento enzimático sustitutivo disponible
| Enfermedad | Enzima deficiente | Herencia | Clínica y/o sistemas afectados | TES (nombre®) |
|---|---|---|---|---|
| Gaucher | β-Glucosidasa ácida (glucocerebrosidasa) | AR | Hepatosplenomegalia Anemia, trombocitopenia Afectación ósea Infiltración pulmonar Neurológica en 2 y 3 | Imiglucerasa (Cerezyme®) 60 UI/kg cada 2 semanas Velaglucerasa (Vpriv®) 60 UI/kg cada 2 semanas |
| Fabry | α-Galactosidasa A | LX | Polineuropatía Hipohidrosis Gastrointestinal Fallo renal Hipertrofia cardíaca | Agalsidasa β (Fabrazyme®)1mg/kg cada 2 semanas Agalsidasa α (Replagal®) 0,2mg/kg cada 2 semanas |
| Pompe | α-glucosidasa ácida | AR | Miopatía progresiva | Alglucosidasa α |
| Glucogenosis II | (maltasa ácida) | Debilidad Cardiopatía Deterioro respiratorio | (Myozyme®) 20mg/kg cada 2 semanas | |
| MPS I | α-Iduronidasa | AR | Hepatoesplenomegalia | Laronidasa |
| Hurler | Disostosis múltiple | (Aldurazyme®) | ||
| Scheie | Dismorfia Hipoacusia Cardiopulmonar Opacidades corneales Cognitivo afectado | 0,58mg/kg cada semana | ||
| MPS II | Iduronato-2-sulfatasa | LX | Similar a MPS I, sin afectación corneal | Idursulfasa |
| Hunter | Piel con pápulas | (Elaprase®) 0,5mg/kg cada semana | ||
| MPS VI | Arilsulfatasa B | AR | Disostosis múltiple | Galsulfasa |
| Maroteaux- Lamy | (N-acetilgalacto-samina-4-sulfatasa) | Hepatosplenomegalia Dismorfia Hipoacusia Afectación cardíaca Afectación corneal No cognitiva | (Naglazyme®) 1mg/kg cada semana |
AR: autosómica recesiva; LX: ligada a X; MPS: mucopolisacaridosis; TES: tratamiento enzimático sustitutivo.
La enfermedad de Gaucher es la EL más prevalente (1/50.000-1/110.000 individuos en Europa occidental). Se origina por el déficit de la enzima β-glucosidasa ácida, que da lugar al cúmulo de glucosilceramida en los macrófagos (células de Gaucher) del hígado, bazo, médula ósea, pulmones y el resto del organismo7. Se hereda de forma autosómica recesiva. Se han descrito más de 300 mutaciones diferentes en el gen GBA (1q21-31), aunque las más frecuentes en la mayoría de poblaciones son N370S (asociada a un riesgo muy bajo de enfermedad neurológica) y L444P (ligada más frecuentemente a las formas neuropáticas de la enfermedad)8.
Aunque en la enfermedad de Gaucher se puede encontrar un «continuo» de síntomas, clínicamente es útil su clasificación, de menor a mayor a gravedad, en: tipo 1 o no neuropático, tipo 3 o neuropático crónico y tipo 2 o neuropático agudo.
La mayoría de pacientes (80-90%) presentan la forma tipo 1, que afecta al sistema reticuloendotelial predominantemente, produciendo infiltración e insuficiencia de la médula ósea, hepatosplenomegalia y enfermedad ósea. En el 70% de los casos la enfermedad se manifiesta en la infancia y suele ser más grave y de progresión más rápida que en la edad adulta. En la primera infancia debe sospecharse ante signos de "enfermedad general", como palidez, astenia, anorexia, retraso del crecimiento, hepatosplenomegalia, con trastornos hematológicos (anemia, trombopenia, leucocitopenia) o alteraciones esqueléticas (dolor, deformidades óseas, necrosis asépticas, fracturas espontáneas). Los primeros síntomas en la adolescencia pueden ser únicamente retraso del crecimiento o de la pubertad.
Los tipos 2 y 3 son casi exclusivos de la infancia, con una frecuencia mucho menor (1/100.000 recién nacidos). La tipo 2 o forma neurológica aguda infantil se inicia precozmente. La hepatosplenomegalia suele aparecer sobre los 3 meses con cuadro simultáneo de regresión psicomotora, parálisis oculomotora, retroflexión del cuello y afectación bulbar. El deterioro suele ser muy rápido y la supervivencia media es de 2 años9. Existen variantes de la enfermedad de Gaucher tipo 2 que se inician en la etapa fetal (hydrops faetalis) o en la etapa neonatal (como bebé colodión o ictiosis congénita). La enfermedad de Gaucher tipo 3 presenta una gran variabilidad clínica.
El TES ha transformado la historia natural de la enfermedad de Gaucher tipo 1, y en los tipos 2 y 3 se utiliza sólo para frenar la evolución o mejorar la calidad de vida. El TPH puede considerarse en formas tipo 3.
La enfermedad de Gaucher tipo 1 fue la primera EL para la que se utilizó TES10. La imiglucerasa (Cerezyme®, Genzyme Corporation) ha mostrado su seguridad y eficacia, corrige las anomalías viscerales y hematológicas rápidamente, mejora o normaliza el crecimiento y revierte la afectación de la médula ósea en muchos pacientes pediátricos, reduciendo la frecuencia de crisis óseas y de fracturas11–13. Para la mejoría de las complicaciones óseas se requiere TES a dosis más altas y durante más tiempo14. No se debe retirar el TES en la infancia ya que se han observado recaídas.
El objetivo del tratamiento en el niño es recuperar al paciente y prevenir manifestaciones futuras. Es muy importante asegurar un estado nutricional adecuado, las medidas de rehabilitación y atención temprana y el tratamiento de las alteraciones esqueléticas.
Recientemente se ha aprobado otra glucocerebrosidasa recombinante, velaglucerasa alfa (Vpriv®, Shire HGT) y está en proceso la taliglucerasa alfa (Protalix®). La disponibilidad de diferentes enzimas para el tratamiento permitirá reevaluar indicaciones, el manejo de los no respondedores y el coste-beneficio de la terapia15.
Lectura rápida
La enfermedad de Fabry es la segunda EL más prevalente, se hereda de forma ligada a X. La sintomatología deriva de la afectación de las células endoteliales vasculares y el músculo liso, dando lugar a isquemia e infarto en el riñón, corazón y cerebro, y otros sistemas. En la infancia, puede iniciarse la sintomatología en la primera década en forma de episodios de dolor neuropático, acroparestesias, manifestaciones gastrointestinales e hipohidrosis. El TES mejora el dolor, la calidad de vida, la sintomatología gastrointestinal, la audición, estabiliza la función renal en pacientes con disfunción renal leve-moderada al inicio, reduce de la masa del ventrículo izquierdo hipertrofiado y revierte la respuesta vascular cerebral anormal.
La enfermedad de Fabry es una enfermedad genética ligada a X que se origina por una actividad deficiente de la enzima lisosomal α-galactosidasa A, lo que ocasiona el cúmulo de glucoesfingolípidos, predominantemente globotriosilcerámido (Gb3). Es la segunda EL más frecuente, con una prevalencia estimada de 1:117.000, aunque un estudio reciente de cribado neonatal sugiere 1/3.100 recién nacidos varones16. Los depósitos de glucoesfingolípidos en los lisosomas afectan mayoritariamente a las células endoteliales vasculares y al músculo liso, dando lugar a isquemia e infarto en el riñón, corazón y cerebro. La afectación de estos órganos mayores se desarrolla a lo largo de la vida, y se manifiesta clínicamente entre la tercera y quinta década de la vida en pacientes no tratados, con insuficiencia renal, hipertrofia ventricular y afectación valvular y accidentes vasculares cerebrales precoces. Las mujeres suelen presentar un cuadro clínico más leve y de inicio más tardío que los varones en general, aunque parecen tener una incidencia mayor de proteinuria en la infancia17.
En la infancia, los afectados de enfermedad de Fabry pueden iniciar su sintomatología en la primera década de la vida en forma de episodios de dolor neuropático, acroparestesias, manifestaciones gastrointestinales y trastornos de la sudoración. Las acroparestesias o dolor neuropático intenso en cualquier localización que se exacerba con el ejercicio o los cambios de temperatura es el síntoma inicial más frecuente. Puede aparecer dolor abdominal posprandial, vómitos o diarrea, e incluso confundirse con una apendicitis o cólico nefrítico. En los estudios retrospectivos, el dolor neuropático se ha descrito en el 63% de los niños y el 46% de las niñas; y las molestias gastrointestinales en el 33% de los niños y el 19% de las niñas. En la adolescencia suelen aparecer lesiones cutáneas en forma de angiomas superficiales, telangiectasias o angioqueratomas. Tienen tendencia a agruparse de forma bilateral y simétrica, en la zona baja del abdomen, genitales y nalgas (distribución «en bañador»). La hipohidrosis es otra manifestación característica, presentando escasa tolerancia al calor y al ejercicio. Las anomalías oculares suelen ser constantes, en forma de opacidades corneales (cornea verticillata) y cataratas de la cápsula posterior (típicas de enfermedad de Fabry). Las alteraciones auditivas como acúfenos e hipoacusia son también frecuentes. En la adolescencia pueden observarse ciertos rasgos faciales, como engrosamiento labial y de los pliegues nasolabiales (50% de los casos). Otras manifestaciones son retraso puberal o talla baja y, ocasionalmente, afectación de órganos mayores con disminución de la tasa de filtración glomerular, proteinuria, hipertrofia ventricular izquierda y anomalías vasculares cerebrales16–18. Para el diagnóstico en mujeres es obligatorio realizar el estudio molecular ya que la actividad enzimática puede tener valores normales al ser heterocigotas.
Actualmente existen 2 preparados comercializados para TES: α-galactosidasa A: agalsidasa α (Replagal®, Shire), y agalsidasa β (Fabrazyme®, Genzyme). Ambos tratamientos han mostrado una eficacia similar para detener y/o enlentecer la progresión de la enfermedad a largo plazo: mejora en las puntuaciones de dolor y calidad de vida, mejoría de la sintomatología gastrointestinal, de la audición, estabilización de la función renal en pacientes con disfunción renal levemoderada al inicio, reducción de la masa del ventrículo izquierdo hipertrofiado y reversión de la respuesta vascular cerebral anormal. El TES se recomienda en cuanto se ha realizado el diagnóstico en adultos varones y en mujeres y niños, actualmente, ante la presencia de manifestaciones clínicas significativas o signos de afectación orgánica mayor18,19.
Enfermedad de PompeLa enfermedad de Pompe, o glucogenosis II, es una enfermedad neuromuscular progresiva originada por una deficiencia de la enzima α-glucosidasa ácida (GAA), que produce un cúmulo de glucógeno en los tejidos originando la sintomatología20. Se hereda de forma autosómica recesiva. Hasta ahora se han descrito más de 200 mutaciones en el gen GAA (17q25). La mutación c32-13T>G es la más frecuente y se asocia a formas de inicio más tardío21, pero la correlación genotipo-fenotipo no es completa. La incidencia en población caucásica es de 1/100.000 nacidos. En Israel o Taiwán es más frecuente (1/40.000 nacidos); y más frecuentes las formas de inicio tardío22. Aunque clínicamente la enfermedad de Pompe puede considerarse también como un espectro continuo, sigue siendo útil su clasificación en la forma infantil precoz (subdividida en clásica y atípica) y forma de inicio tardío, a partir del año de vida (subdividida en juvenil y del adulto). Todas presentan una miopatía progresiva. Cuando la actividad enzimática es inferior al 1% la alteración de la función muscular es tan rápida e intensa que produce una enfermedad de inicio rápido y agresivo, dando la forma clásica, la más frecuente, en donde además se afecta el músculo cardíaco produciendo a cardiomegalia.
Los niños con enfermedad de Pompe clásica presentan problemas de succión y alimentación desde el nacimiento dando lugar a escasa ganancia ponderal y a dificultades respiratorias que les predispone a infecciones respiratorias graves, antes de los 3–5 meses23. Tienen hipotonía con ausencia de reflejos osteotendinosos. La mayoría de los casos tienen cardiomegalia antes de los 6 meses por una cardiomiopatía hipertrófica, y alteración de la conducción cardíaca con complejos QRS grandes y PR cortos típicos en el ECG. Más del 50% tienen macroglosia y hepatomegalia. Analíticamente se observa un aumento de la creatincinasa (CPK) y de las transaminasas (alanino y aminotransferasa), por afectación muscular y hepática20. La electromiografía (EMG) muestra patrón miopático y ocasionalmente puede aparecer patrón miotónico. La enfermedad de Pompe precoz atípica tiene una evolución más lenta, sin cardiopatía o con leve hipertrofia ventricular y con supervivencia hasta los dos años. La edad media para precisar ventilación mecánica, sin tratamiento, es a los 5,9 meses y el fallecimiento a una edad media de 8,7 meses; menos de 25% superan el año de vida y el 7,1% viven más de 2 años24.
La forma juvenil de la enfermedad de Pompe, de inicio tardío, puede presentarse en cualquier momento tras el año de vida y se caracteriza por retraso motor, hipotonía y debilidad muscular proximal. Los músculos respiratorios se afectan precozmente y la muerte se produce por fallo respiratorio en ausencia de miocardiopatía. La forma adulta, menos frecuente, se inicia entre la segunda y cuarta década de la vida (edad media de inicio a los 24 años), incluso después de los 60 años y cursa como una miopatía lentamente progresiva. El fallo respiratorio suele también desencadenar la muerte en estos casos.
Precisa un abordaje mutidisciplinario para el tratamiento, con especial atención en este caso al tratamiento sintomático respiratorio, cardiológico, dietético y fisioterápico25. En 2006 fue aprobado el uso comercial de la enzima recombinante (Myozyme®, Genzyme Corporation) en Europa y EE.UU. En la actualidad hay más de 300 casos tratados en el mundo. Se ha mostrado eficaz en el aumento de la supervivencia y la mejora de las funciones cardíaca, respiratoria y motora26,27; se reduce el riesgo de ventilación invasiva en un 58% y el riesgo de muerte en un 79%28. Se sugiere en un estudio reciente que los resultados observados no son tan buenos a largo plazo si el inicio del tratamiento es tardío y la situación clínica de los pacientes es más grave29.
Lectura rápida
La enfermedad de Pompe se hereda de forma autosómica recesiva. Presenta una afectación neuromuscular progresiva. Los niños con EP clásica presentan hipotonía con ausencia de reflejos osteotendinosos, cardiomegalia y alteración de la conducción cardíaca. El aumento de creatinfosfocinasa (CPK) y de las transaminasas, junto con el electromiograma (EMG) orientan al diagnóstico. El TES aumenta la supervivencia y mejora las funciones cardíaca, respiratoria y motora.
Las mucopolisacaridosis (MPS) I y VI se heredan de forma autosómica recesiva y la MPS II de forma ligada a X. Presentan un espectro continuo de síntomas desde formas graves a leves. Suelen ser asintomáticos al nacimiento, con aparición progresiva de la clínica.
La MPS I es una enfermedad lisosomal autosómica recesiva originada por el déficit de la enzima α-iduronidasa que ocasiona la acumulación progresiva de GAG, dermatán y heparán sulfato30. Se han descrito más de 100 mutaciones diferentes en el gen responsable (4p16). Las mutaciones W402X, Q70X y P533R son responsables de la mitad de los alelos en la población de origen europeo31. Su prevalencia general se estima en 1,07/100.000 nacimientos32, aunque la incidencia en Irlanda es mucho mayor y la más alta conocida hasta la fecha, 1/26.206 recién nacidos33. Clásicamente se describen 3 formas clínicas. La forma clínica más grave, de progresión más rápida, o síndrome de Hurler (MPS IH); la atenuada, de progresión más lenta, o síndrome de Scheie (MPS IS) y la intermedia denominada síndrome de Hurler-Scheie (MPS IHS). Hoy día se prefiere clasificarla en 2 grupos, la forma grave (IH) y la forma atenuada (IS y IHS)34.
Los niños con MPS IH suelen ser asintomáticos al nacimiento y progresivamente va apareciendo hepatosplenomegalia, rasgos faciales toscos, opacificación corneal, displasia esquelética (disostosis múltiple), baja talla, hipoacusia, retraso mental progresivo, afectación cardíaca (hipertrofia septal y valvulopatía) y fallo cardiorrespiratorio progresivo. Su crecimiento se va enlenteciendo a partir del primer año de vida, y su retraso psicomotor es evidente durante el segundo año de vida, con regresión motora y prácticamente ausencia del lenguaje. La disostosis múltiple característica se constata radiológicamente (fig. 1) por engrosamiento de huesos craneales, silla turca agrandada en forma de J, deformidades de vértebras lumbares en forma de gancho, cifoescoliosis, acortamiento de huesos largos, pelvis hipoplásicas con cabezas femorales pequeñas y huesos de manos y pies acortados y de forma trapezoidal30. Sin tratamiento, los pacientes con MPS IH tienen una supervivencia de 6,8 años32. Las formas más leves (atenuada e intermedia) pueden no desarrollar síntomas hasta los 3–10 años, se caracterizan por rigidez articular, valvulopatía aórtica y opacidad corneal, pero la inteligencia y la talla suelen ser normales y pueden sobrevivir hasta la adolescencia o edad adulta.
 Deformidades de vértebras lumbares en forma de gancho y cifoescoliosis; B engrosamiento de huesos craneales y silla turca agrandada en forma de J; C huesos de manos cortos y anchos.")
Desde la década de los años 1980 algunos pacientes con MPS I han recibido TPH, sobre todo alogénico de médula ósea. Un trasplante con éxito puede preservar, si se realiza muy precozmente (por debajo de los 2 años), las funciones neurocognitivas y mejorar algunos aspectos de la enfermedad somática (hepatosplenomegalia, fenotipo facial, rigidez articular, enfermedad cardíaca y apnea del sueño), aunque tiene muy poco impacto en la enfermedad ósea y un efecto muy variable a nivel ocular. Se han publicado algunos trabajos en los que el uso previo de TES mejora las tasas de supervivencia y de implante35. El TES con laronidasa (Aldurayme®; Genzyme Corporation) fue aprobado en 2003 y más de 100 pacientes han sido tratados hasta la fecha. Sus beneficios clínicos incluyen disminución de la hepatomegalia, mejoría de la función respiratoria, mejoría de la deambulación, aumento de la amplitud de los movimientos articulares, disminución de la hipertrofia ventricular izquierda y disminución de la excreción urinaria de GAG; además de una muy buena tolerancia36–40.
Lectura rápida
En la MPS I y II aparece hepatosplenomegalia, rasgos faciales toscos, displasia esquelética (disostosis múltiple), baja talla, hipoacusia, retraso mental progresivo, afectación cardíaca (hipertrofia septal y valvulopatía) y fallo cardiorrespiratorio progresivo. Los beneficios del TES incluyen disminución de la excreción de glucosaminoglucanos, reducción de la hepatomegalia, aumento de la capacidad vital forzada respiratoria, mejora de la deambulación y reducción de la masa ventricular izquierda.
En la MPS VI clínicamente destaca la dismorfia y la displasia esquelética con talla corta y degeneración articular, sin deficiencia intelectual. El TES mejora el crecimiento, el desarrollo puberal y la función pulmonar.

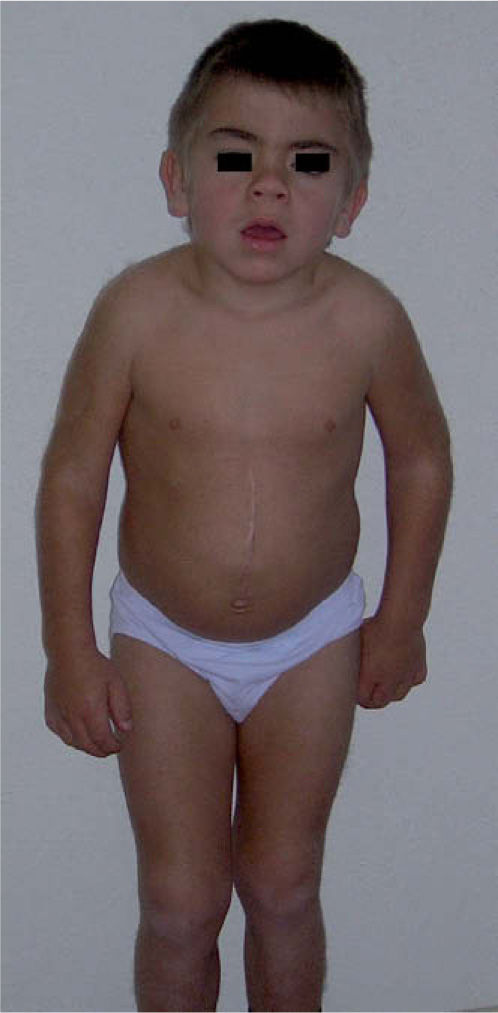
La MPS II se produce por un déficit de la enzima iduronato-2-sulfatasa (I2S) ocasionado por mutaciones en el gen IDS (Xq28). Más de 300 mutaciones se han descrito hasta la fecha. Es la única MPS que se transmite con un patrón ligado a X y su prevalencia estimada en los recién nacidos varones es de 1/170.000; también se ha descrito MPS II en algunas mujeres heterocigotas41. Actualmente la clasificación clínica en forma grave y leve o atenuada se considera muy simplista y es mejor considerarla como un espectro continuo de fenotipos entre los dos extremos. Sus rasgos clínicos son muy parecidos a la MPS I, con tosquedad facial, macrocefalia, hirsutismo, cifoescoliosis, hepatosplenomegalia, hernias abdominales y/o inguinales y talla baja (figs. 2 y 3). Presenta pápulas cutáneas características, además de afectación cardíaca (sobre todo valvular), esquelética, respiratoria y neurológica progresivas42–44. Sin tratamiento, la muerte suele ocurrir en torno a la adolescencia en la forma más grave de la enfermedad, mientras que las formas leves, sin afectación neurológica, pueden sobrevivir hasta la época adulta44. El TES con I2S recombinante (Elaprase®, Shire HGT) fue comercializado en Europa en 2007, tras comprobarse su eficacia y seguridad en los ensayos clínicos previos44. De forma prospectiva y desde entonces también se ha confirmado su beneficio a través del Hunter Outcome Survey (HOS), registro multicéntrico e internacional de datos de MPS II iniciado en 2005 para mejorar el conocimiento de la historia natural de la enfermedad y vigilar la seguridad y eficacia del TES. Hasta enero de 2010 se han seguido 259 pacientes europeos con TES, en los que se ha observado de forma significativa disminución de la excreción de GAG, reducción de la hepatomegalia, aumento de la capacidad vital forzada respiratoria, mejora de la deambulación y reducción de la masa ventricular izquierda45. Recientemente se ha publicado una seguridad y eficacia similar del TES en niños españoles menores de 5 años tratados precozmente46.
Mucopolisacaridosis VI o síndrome de Maroteaux-Lamy

La mucopolisacaridosis VI (MPS VI) es una enfermedad autosómica recesiva, causada por mutaciones en el gen de la arilsulfatasa B (ARSB) localizado en 5q13-14. La prevalencia media de MPS VI es de 1/238.000 nacidos. En la población inmigrante turca en Alemania es mucho más frecuente (1/43.261nacidos)47.
La sintomatología más llamativa está determinada por la dismorfia y la displasia esquelética características que incluyen talla corta, disostosis múltiple y degeneración articular. Las formas rápidamente progresivas pueden tener inicio desde el nacimiento y la muerte puede ocurrir antes de la segunda década. Las formas más lentamente progresivas pueden tener desarrollo normal en los primeros meses y los pacientes vivir hasta la quinta década. Otros problemas frecuentes son: afectación de las válvulas cardíacas, alteración de la función pulmonar, hepatosplenomegalia, sinusitis, otitis media, pérdida auditiva, apneas del sueño, opacidades corneales, síndrome del túnel carpiano y hernias inguinales y umbilicales. En general, no hay deficiencia intelectual, pero puede haber otros problemas neurológicos por compresión medular, inestabilidad de la columna cervical, engrosamiento meníngeo o estenosis óseas, hidrocefalia comunicante, atrofia del nervio óptico y ceguera48.
Hasta hace poco, el tratamiento sintomático y el TPH eran los únicos tratamientos disponibles. El TES se realiza con galsufasa (Naglazyme®, Bio-Marin), autorizada en Europa en 2006, y mejora el crecimiento, el desarrollo puberal y la función pulmonar49. Su efectividad es mayor si se utiliza precozmente e incluso en estadios preclínicos50.







