




Special issue on hepatocellular carcinoma (HCC) and hepatitis B and C as its main causes worldwide
Más datosHepatitis B Virus is classified into ten different genotypes (A- J). Genotypes F and H cluster apart from others in phylogenetic trees and is particularly frequent in the Americas. The aim of this study was to sequence complete genomes of samples of HBV genotypes F and H from Brazil and Mexico using next generation sequencing (NGS) and to study relevant characteristics for the disease associated with this virus.
Materials and methodsNinety plasma samples with detectable HBV DNA belonging to the F (n=59) and H (n=31) genotypes were submitted to amplification of the complete HBV genome by three different methologies. Data analysis was developed using bioinformatics tools for quality assurance and comprehensive coverage of the genome. Sequences were aligned with reference sequences for subgenotyping and detecting variants in relevant positions. A phylogenetical tree was constructed using Bayesian methods.
ResultsHBV genome of 31 samples were amplified and 18 of them were sequenced (HBV/F=16 and HBV/H=2). One genotype F sample was co-infected with the F1b and F3 subgenotypes, while the other samples were all F2a subgenotype. Two genotype H samples clustered with other Mexican sequences. The main variants observed were found in preS and S genes (7/18) and mutations in the precore/core region (11/18).
ConclusionsA NGS methodology was applied to F and H genotypes samples from Mexico and Brazil to fully characterize their sequences. This methodology will be relevant for clinical and epidemiological studies of hepatitis B in Latin America
Hepatitis B virus (HBV) belongs to the Hepadnaviridae family, which also includes other HBV-related viruses identified in different species. HBV is classified into 10 different genotypes [1].
Genotypes A and D currently have a worldwide distribution, but are more likely to originate from the western region of the Old World. Genotype A is further divided into different subgenotypes, from A1 to A7, which have a characteristic distribution among different populations from different geographic regions. Genotype D is divided into 9 different subgenotypes, characteristic of different populations from Asia, Africa, Europe, and Oceania [2].
Genotypes B and C are common in Asia, from Indonesia in the South to East Russia in the North, and the Philippines, China and Japan in the East. Genotype B is also common in some native populations of North America (mainly the Inuit), in Alaska, northern Canada and Greenland. Genotype B was classified into 9 different subgenotypes, which are distributed among different populations that inhabit these regions. Genotype C is found in the same regions of Asia, but it is not common in ancient population groups of North American countries, but it is found in autochthonous populations of countries in Oceania and is further classified into 16 different subgenotypes. Genotype E is native to Africa and predominates in the Western region of that continent, being rarely found outside this location [3].
Genotype G was originally found in men who have sex with men and other publications have shown its presence in Germany, France, Spain, USA, Mexico, Colombia and Brazil. In fact, this genotype has been found infrequently in different populations around the world and it seems to be phylogenetically closer to the genotype A, with an insertion in the preC region and other alterations in the preS region, the latter curiously shared with the genotype E [4].
Two other genotypes have also been described recently, genotype I, found in Vietnam and Laos, and genotype J, found in Indonesia, which is a likely recombinant with a virus found in Gibbons [3].
A classification of HBV genotypes into three hierarchical groups of higher order was proposed: group I, composed of genotypes A-E and G; group II, comprising the F and H genotypes; and a hypothetical group III. The current G genotype is postulated to be a recombinant with the non-polymerase region of the group III virus and the polymerase gene of an ancestral virus belonging to the group I virus. Although this classification is not widely used, it has the advantage of isolating genotypes F and H, which are genotypes found mainly in Native American populations [5].
Genotype F is characteristic of America, being the only genotype found among isolated indigenous populations and widely distributed in regions with a strong contribution of this racial group in its formation. Genotype F strains isolated in different geographic regions are classified into 4 subgenotypes: F1, whose clade is subdivided into F1a (Costa Rica, El Salvador) and F1b (Argentina, Chile, Alaska), F2, which also has a subdivision into F2a (Venezuela, Brazil, Argentina, Nicaragua) and F2b (Venezuela, Martinique), F3 (Venezuela, Colombia, Panama) and F4 (Argentina, Brazil, Bolivia) [6,7].
The aim of this study was to sequence the complete genome of HBV genotype F and H samples from Brazil and Mexico using the next-generation sequencing methodology. A secondary objective was to study relevant characteristics for the understanding of diseases associated with this virus from the data obtained by the analysis of the generated sequences:
1Materials and methods1.1SamplesFor this study, 90 plasma samples obtained from patients infected with HBV genotype F (n=59) or genotype H (n=31) were included. Genotype F samples were obtained from patients from different Brazilian states: Pará (n=12), Maranhão (n=6), Minas Gerais (n=4), São Paulo (n=36), and Rio Grande do Sul (n=1). The samples belonging to genotype H were obtained from patients from Guadalajara, Mexico.
All 59 samples of genotype F were HBsAg positive using standard routine immunoenzimatic assays. Their viral load was determined using Abbott RealTime HBV Viral Load Assay (Northern Chicago, IL, USA) and ranged between 2.65 log and 8.95 log, with most samples quantified with a viral load below log 5. Of the 31 samples of genotype H used in the study, 15 were HBsAg positive and for the others this information was not available.
1.2Viral DNA extractionDNA from each sample was extracted from 200 μl of plasma using the QiaAmp DNA mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
1.3Selection of primers and strategies for amplification of the complete HBV genomeThe complete genome of the hepatitis B virus was amplified by polymerase chain reaction (PCR) using three protocols (A, B and C), with different combinations of primers in order to amplify fragments that encompass the entire nucleotide sequence of the same.
In protocol A, primers P1 and P2 described by Günther et al. [8] were utilized, which anneal at positions in the genome that allow the amplification of the 3215 bp of the HBV genome in a single reaction.
Protocol B consisted of performing three PCRs using different primer combinations to amplify three overlapping fragments: P1 and P194 were used to generate the first fragment of 1611 bp; PS3076F and P1196R to generate the second 1335 bp fragment; and 5′LAM5 and P2 to generate the third 1210 bp fragment [9].
In protocol C, the complete HBV genome was amplified using primers that generate 2 overlapping fragments using nested PCR to improve the sensitivity of the reaction for amplification of the DNA of interest, thus enabling greater success in the amplification of samples with low levels of viral load. The primers used in this last strategy were chosen from the study carried out by Wang et al. [12] to amplify and sequence with high accuracy and sensitivity samples from HBV-infected patients with low viral load. For amplification of the first fragment, primers P1 and AR1 were used to perform the first PCR, and P1 and AR2 in the nested PCR reaction, generating a 2092 bp fragment. For amplification of the second fragment, primers P2 and AF1 were used in the first reaction, and P2 and AF2 in the nested PCR reaction, resulting in a fragment of 1320 bp. All primers chosen in the protocols performed and their respective sequence and position in the genome are listed in Table 1.
Primers utilized in the PCR methodologies utilized for the amplification of the whole HBV genome.
| Primer | Sequence 5’-3’ | Position | References |
|---|---|---|---|
| P1 | CCGGAAAGCTTGAGCTCTTC | 1821-1841 | 8 |
| P2 | AAAAAGTTGCATGGTGCTGG | 1825-1806 | 8 |
| P194 | AAACCCCGCCTGTAACACGA | 211-192 | 9 |
| PS3076F | TGGGGTGGAGCCCTCAG | 3079-3095 | 10 |
| P1193R | GCGTCAGCAAACACTTGGCA | 1193-1174 | 9 |
| 5’LAM5 | TGCRYYTGTATTCCCATCCCATC | 593-615 | 11 |
| AR1 | ACAGTGGGGGAAAGC | 759–745 | 12 |
| AR2 | AGAAACGGRCTGAGGC | 702–687 | 12 |
| AF1 | GTCTGCGGCGTTTTATC | 419–435 | 12 |
| AF2 | TGCCCGTTTGTCCTCTA | 503–519 | 12 |
For the amplification reactions, the enzyme Q5® Hot Start High-Fidelity DNA Polymerase (New England Biolabs, Massachusetts, USA) was used in all protocols. This enzyme was chosen as it has a low error insertion rate (approximately 280 times lower than Taq DNA polymerase). Half microliter of the enzyme was used in a mix containing 10 μl of 5X Q5 Reaction Buffer); 26.5 µl of H2O; 1 μl of dNTPs at 10 mM and 1 μl of the protocol-specific primers at a concentration of 30 pmol/μl. Subsequently, samples were subjected to repeated cycles of temperature variation in the Mastercycler proS thermocycler (Eppendorf AG, Hamburg, Germany) for amplification of each target.
For the reactions using protocol A, an initial denaturation step was performed at 98°C for 30 s followed by 40 cycles at 98°C for 10 s, 64°C for 30 s, 72°C for 2 min. At the end of the amplification cycles, an extension step at 72 ºC for 2 min was added to fill in any incomplete polymerization.
The reactions for protocol B were subjected to the following cycles of temperature: 98°C for 5 min followed by 34 cycles of 98°C for 10 s, 61°C for 30 s, 72°C for 50 s; and a final step at 72°C for 5 min.
The cycling conditions applied to the reactions assembled with protocol C primers were as follows: 98°C for 2 min followed by 30 cycles of 98°C for 10 s, 58°C (for the first round of amplification) and 53.7°C (for nested PCR reaction) for 30 s; 72°C for 1 min and 30 s; and a final step at 72°C for 5 min.
The amplified product was identified by agarose gel electrophoresis (4 μl of the final PCR product mixed with 1 μl of the 10X Bluejuice™ Gel Loading Buffer (InvitrogenTM Life Technologies, Carlsbad, CA, USA) and the mixture was applied to a 1% agarose gel prepared with 1 X TAE buffer. and 7 µl of SYBR Safe.
1.5Preparation of libraries for next-generation sequencing and sequencing of amplified HBV genomesDNA library was prepared from the amplicons generated in the PCR reactions, using the Nextera XT DNA Sample Preparation kit, following the procedures established by the manufacturer. After quantifying the PCR products using the Qubit fluorimetric method (Qubit 4 Fluorometer; Thermo Fisher Scientific, Waltham, Massachusetts, USA), the concentration of all PCR samples was adjusted to 0.8 ng/μl, and then performed the DNA fragmentation and tagmentation steps. Samples were then submitted to amplification step for insertion of adapters and were then DNA purified using AMPure XP magnetic beads (Beckman Coulter; Life Sciences Division Headquarters; Indianapolis, USA). Samples were purified, let all at 2 nM of final concentration and denatured using a 0.2 N NaOH solution (v/v). The ready-made libraries were pipetted on a flow cell and then the sequencing process was started using the MiSeq platform (Illumina Inc., San Diego, CA, U.S.A.).
1.6Analysis of HBV Full Genome SequencesAfter sequencing, the raw data underwent a quality analysis using the FastQC and After QC tools (Babraham Bioinformatics, Babraham Hall House, Babraham, Cambridge CB22 3AT, United Kingdom) to obtain basic library statistics such as: file type, format, total sequences, filtered sequences, sequence length and percentage of guanine + cytosine (GC) content of each base in each generated sequence [13]. Quality score parameters per sequence, sequence quality per base, curve quality, curve discontinuity, GC content per sequence, N content per base and sequence length distribution were also verified. To clean up noise from the sequences of each library, a trimming was performed, followed by the removal of short reads with bad overlap, removal of reads with low sequence quality, with poly-X or excess N.
The contigs were generated with the SPAdes program [14], using different sizes of k-mer and selecting the result with the highest contigs. First, the contigs were compared to sequences from a local database cured only for HBV, using the BLAST (Basic Local Alignment Search Tool) in order to identify the similarity with the bank sequences, allowing the identification of genotype and subgenotype. To assemble the final sequence, we used multiple contigs from each sample, according to the local BLAST.
1.7Phylogenetic AnalysisThe generated sequences were aligned with reference sequences from each HBV genotype and subgenotype, including several sequences from the F and H genotypes isolated in Latin America, using the Clustal_W integrated with the BioEdit [15] software. Subsequently, the alignment was submitted to phylogenetic analysis using the BEAST v.1.8.3 [16] program with 10,000,000 steps of the Markov Monte Carlo Chain (MCMC) sampling every 1,000 steps, and rejecting the first 1,000,000 steps as burn-in. The FigTree v1.4.2 program (http://tree.bio.ed.ac.uk/software/figtree/) was used to view and edit the phylogenetic trees. The analysis was performed by the Bayesian method from 186 sequences of the complete HBV genome from different genotypes (167 from GenBank and 19 from this study).
In the call of variants, the Freebayes tool [17] and the functional annotation with the SnpEff program [18] were used, which are capable of analyzing and annotating thousands of variants and predicting their possible genetic effects. The quality filter was defined considering a Bonferroni correction [19] starting from a cut-off of Q=30, that is, a false positive rate of 1/1000, where ∼3215 sites were evaluated with a cut- off corrected from 1.8e−8, which corresponds to q=77. The main high impact variants found in the HBV literature on the pre-core and basal promoter of the core genes, preS, S, X and P gene were placed as preference in the analysis filter. All variants were raised in comparison with the sequence KJ843191, which is an F2a genotype with the complete genome deposited in GenBank.
In the annotation of mutations, variants stipulated as high impact with loss of function were described. Variants in the pre-core, core, pre S, gene X and polymerase regions were inspected.
2Results2.1Amplification of complete HBV genomesAll 90 samples (HBV/F=59; HBV/H=31) were processed using one of the HBV complete genome amplification protocols described. Using protocol A, 18 samples belonging to genotype F and none of the genotype H samples were amplified.
After the result of this first protocol, to avoid wasting reagents and samples, the next tests were divided into stages. Initially, of the 41 genotype F samples that were not amplified using protocol A, 35 samples of genotype F were separated and subjected to amplification using the protocol B primers (P1.2/P194; PS3076F/P1196R and 5′LAM5/P2.2). Through this strategy, it was possible to amplify the complete HBV genome in 5 samples from the three overlapping fragments obtained.
For the amplification of the 36 genotype F samples that did not show bands on the gel in the previous protocols, a nested PCR protocol was performed for amplification of two overlapping fragments (protocol C). The samples were separated and tested, and it was possible to amplify the complete genome of 5 samples.
For genotype H, the 22 samples that did not amplify with protocol A and the 9 samples that have not yet been processed were tested using protocol C and 3 had the complete HBV genome amplified.
2.2Next Generation SequencingFinally, it was possible to amplify the complete HBV genome from 31 samples of the 90 tested (genotype F = 28; genotype H = 3). All of them were submitted to next-generation sequencing and good quality sequences were only obtained in 16 and 2 samples among the 31 and 3 samples with genotypes F and H, respectively.
As seen in Table 2, these samples have at least 87% (most of them 100%) of coverage with mean and median coverage levels ranging from 215.4 - 32456.9 (mean) and 228-33332 (median).
Percentage and coverage from the HBV whole genome sequences obtained.
The genomes were assembled and their identification was based on the results of the local alignment that compared the sequences produced with the non-redundant nucleotide sequence bank from https://blast.ncbi.nlm.nih.gov/Blast.cgi. Table 3 shows the results obtained from all the samples. All the HBV complete genome sequences have more than 98.7% identity with a reference sequence from a specific genotype or subgenotype, confirming the genotypes previously identified in the partial genome sequencing and identifying the subgenotypes. No recombination events were identified in the characterized HBV/F and HBV/H sequences.
Identity and genotype/subgenotypes of the sequences here obtained compared to the NCBI databank compared to BLAST.
CO20 is shown twice as it was possible to identify two different sequences.
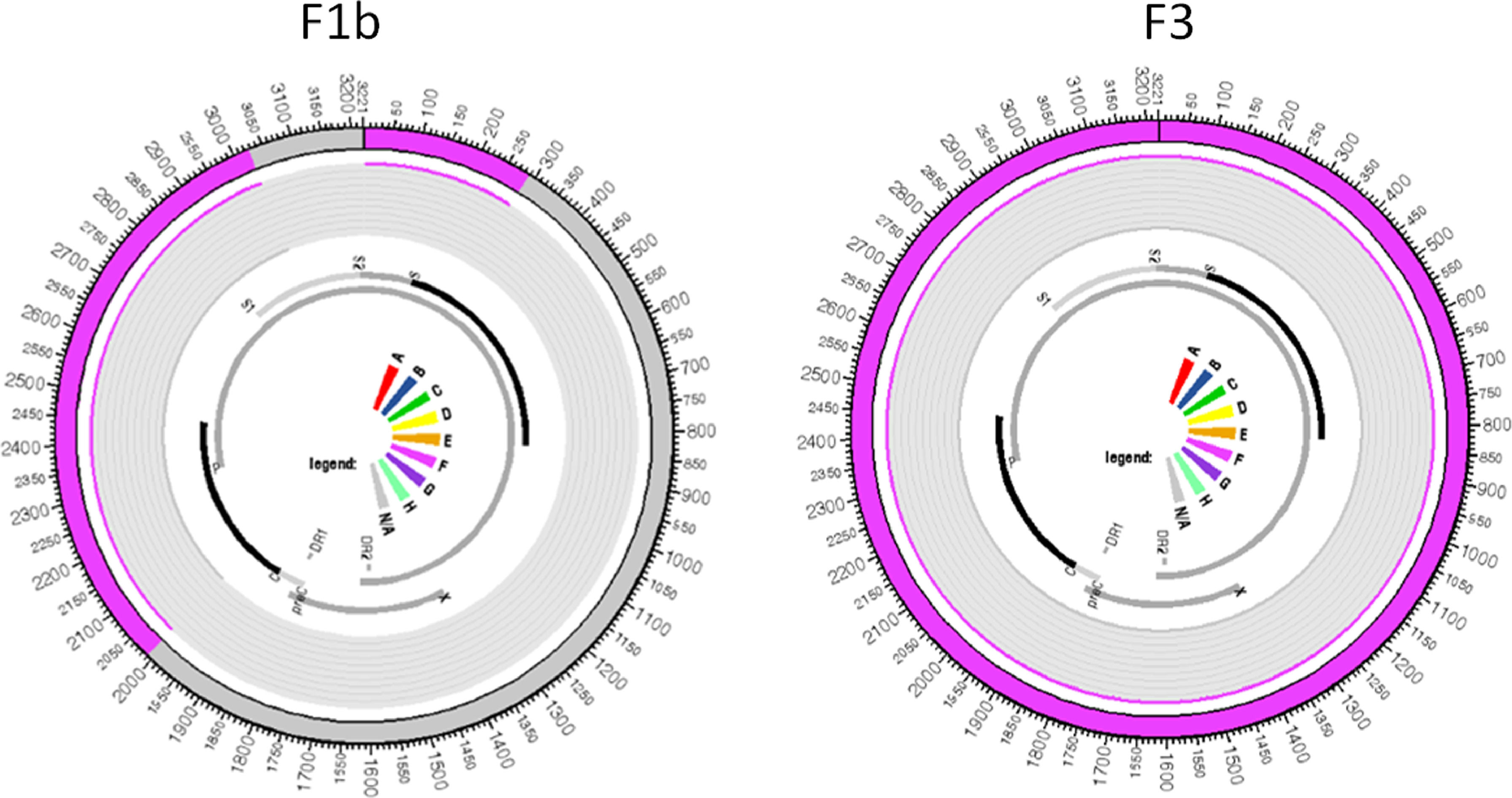
All samples belonging to the F genotype were classified as subgenotype F2a, with the exception of the CO20 sample in which reads belonging to two different subgenotypes were identified, where the largest read showed 99.8% similarity to the reference sequence of the subgenotype F3 and the other read generated from smaller size showed 99.2% similarity to a complete reference sequence of the subgenotype F1b. These two reads aligned to the same portion of the HBV genome in their respective reference sequences: the first contig aligned to practically the entire genome of the subgenotype F3 reference, from 2 to 3215, with 98.87% identity; the second contig showed an identity of 99.61% with the nucleotide regions 2 to 286 and 2013 to 3019 of an F1b genotype reference sequence, which evidences the co-infection with these two subgenotypes (Figure 1).
, and the largest contig aligned to the F3 reference.")
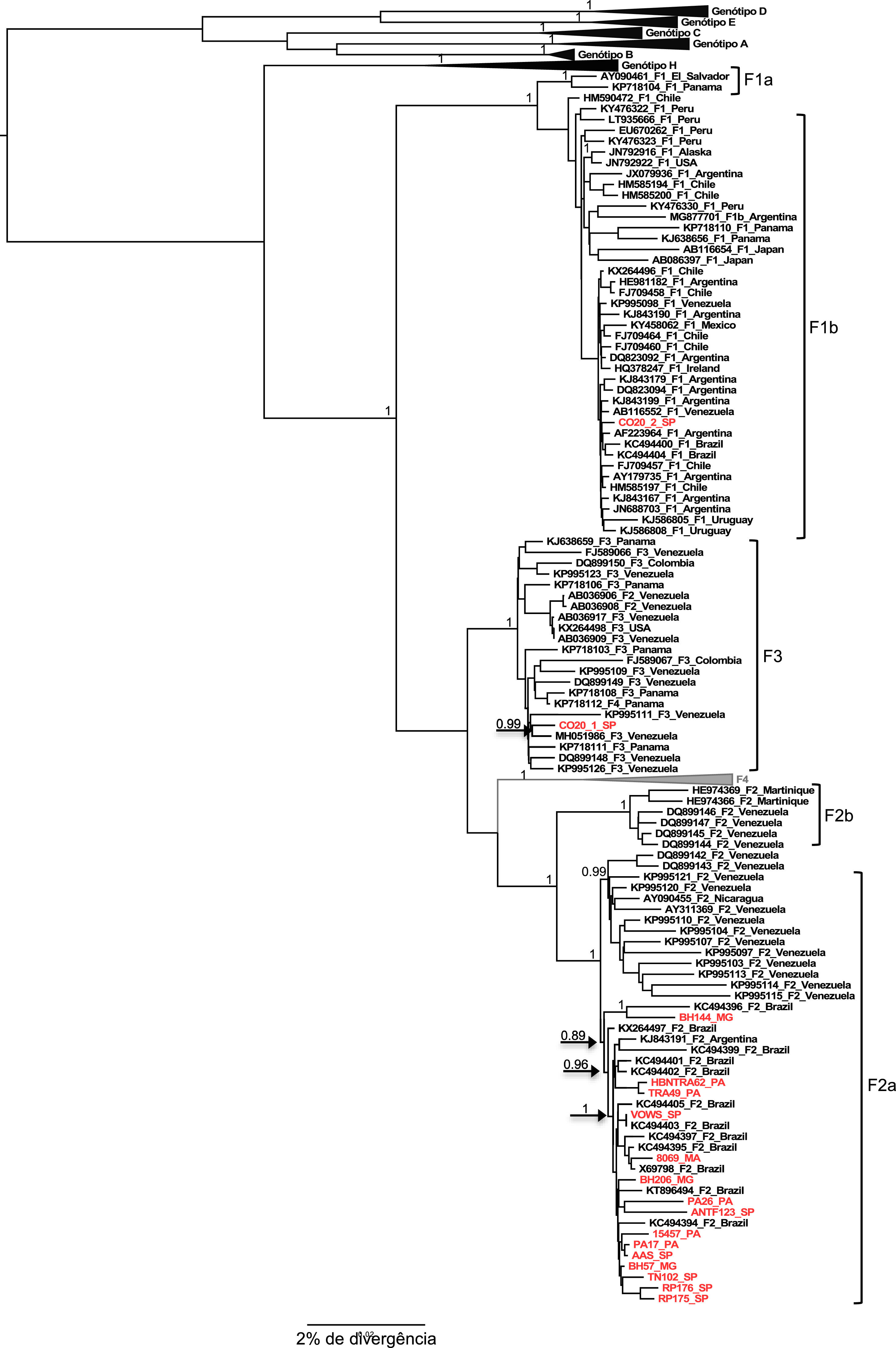
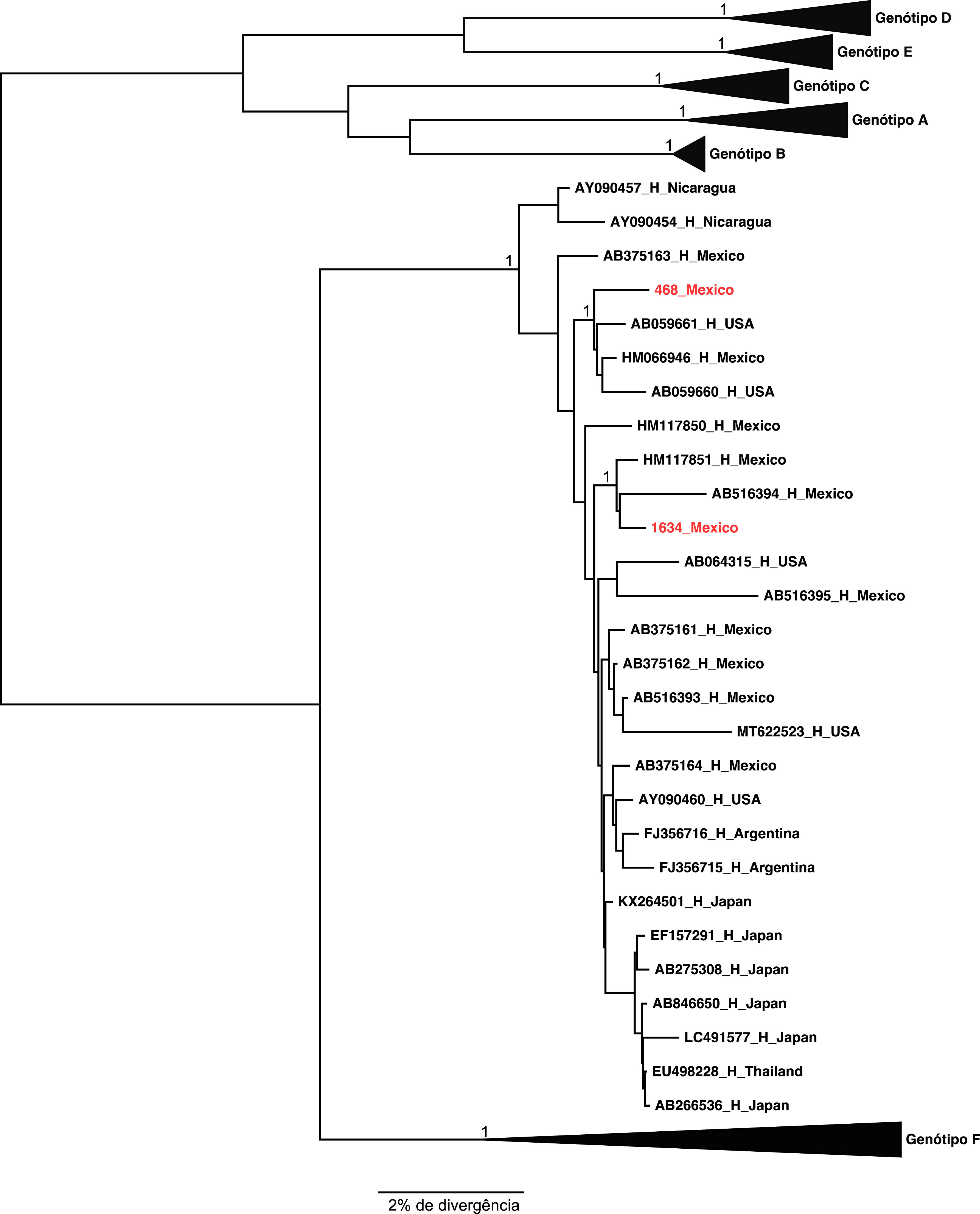
The two samples belonging to genotype H grouped, as expected, with the other reference sequences isolated also in Mexico. These results were confirmed by the phylogenetical tree (Figures 2 and 3).


The result of the phylogeny of the F2a sequences characterized in this study points to the formation of a specific geographic grouping with F2a sequences circulating in Brazil, but the formation of clades according to the region of origin of the sample within the country is not observed, as isolated sequences of cases from the different regions are intermingled.
Regarding the F1b and F3 sequences isolated from the CO20 sample, it is observed that both group together closely with sequences of the respective subgenotypes isolated in Venezuela (Figure 2), which suggests a common origin of these strains identified in the same patient.
2.4Viral variantsS
Strains with premature stop codons in the S gene (sC69STOP, sW196STOP or sL216STOP) were identified in 5 samples, genotypes F=3 and H=2, making up 12.5 to 70% of the viral population. In 9 samples, genotypes F=7 and H=2, HBV strains with mutations within the major hydrophilic region of HBsAg were identified, and only two of the observed mutations (sQ129H and sS140T) were already associated with escape of anti- HBs. HBV strains with the sQ129H or sS140T mutations were identified in 6 samples (8096, BH144, BH57, PA17, RP176, 468).
2.5Pre SA premature stop codon at amino acid 85 of the pre-S1 region (ps1L85STOP) was observed in sample BH57 (genotype F2a) and 40% of the viral population had this change. Variants with mutation in the pre-S2 gene initiation codon was observed in sample TN102 (genotype F2a) with a frequency of 98%. Variants with deletion in the pre-S2 gene were observed in sample TRA62 (genotype F2a) with a frequency of 55% and composing 94.6% of the viral population of sample 1634 (genotype H).
2.6PolymeraseMutations in 16 different amino acids encoded by this genomic region were identified in 9 samples (Genotype F=7 and H=2), and none of them conferred proven resistance to AN, only two have been associated with a potential impact on treatment response with Adefovir (rtS85A and rtS219A). Among these, only rtS219A was observed in the majority viral population (frequency of 88.8 to 94%), in the ANT123 sample.
2.7Basal core promoter (BCP), pre core, core and XMutations in the BCP were observed: A1762T in 4 samples (15457, ANT123, TN102, 1634) with a frequency of 78.3 to 99.5% and G1764A (1 sample: PA26) constituting 98.1% of the viral population. No sample shared these mutations concomitantly. Nucleotide substitutions at positions 1762 and 1764 cited above lead to mutations in protein X at positions 130 (xL130M) and 131 (xV131I), respectively. Another HBx mutation associated with clinical impact was observed at amino acid 127 (xI127L) in the HBV F2a genome isolated from the RP17 sample.
Mutations (substitutions or deletions) in the pre-core gene initiation codon were found in 10 samples (Genotype F=8 and Genotype H=2), in all cases representing the dominant viral population (87.5 to 100%).
3DiscussionThe amplification of the complete HBV genome has been a challenge for researchers since 1995 when Günther et al. [8] proposed in their study a pioneering way of amplifying the complete genome of the virus using a pair of primers. In this study, the idea of using only one pair of primers to amplify 3200 base pairs at once was applied and the technique of cloning the product of the first PCR into plasmids was also used to amplify the DNA segment of the virus inserted into them. The next studies carried out in the following decades on the subject continue to have some difficulty in reproducing, and most of the studies characterize a small number of samples, as we can see, for example, by the studies by Sasha et al. [20], in which 6 primers were utilized for amplification of the complete HBV genome from only one sample; by Spitz et al. [21] who used the same Günther method of cloning in plasmids to amplify only one sample; by Fonseca et al. [22] who used 10 primers to amplify three samples; and even the study that characterized the G genotype by Stuvyer et al. [10], whom, unlike the others, characterized the complete genome of the samples by amplifying with a greater number of primers, using 21 pairs in a nested and semi-nested reaction PCR.
In the present work, even in the attempt to optimize the amplification reaction of the complete HBV genome, in addition to having a smaller number of primer pairs available, we utilized the Q5 high-fidelity enzyme, the same proposed by Wang et al. [12] as the chosen for HBV amplification, since in addition to having an error rate less than 10 times compared to other commonly used enzymes, it also reduces the time needed to carry out the amplification of a long DNA fragment.
Another important issue for the successful amplification of the virus is related to the viral load present in the investigated sample: the higher the viral load in the sample, the higher the yield of amplified products to be sequenced and the easier to assemble a reliable complete genome. The HBV genotype H is in several studies related to low viral load, as demonstrated by Sozzi et al. [23] in the study where it is proven through cell culture tests that this genotype has lower rates of replication and expression of HBsAg and HBeAg compared to other genotypes.
Characterization of the complete genome of a virus can be useful for clinical diagnosis, either by revealing the agent involved in the infection or by identifying clinically important genetic variations. As an example, many studies have demonstrated that, precore/core promoter mutations and S gene mutations, including pre-S deletions, are related to different antigen expression level and immunogenicity, and related to progressive liver disease and risk of developing HCC [24].
There are reports of failure for vaccination due to mutations in the S gene, since vaccines generate responses to epitopes located in the HBsAg protein. In 2011, Tackle et al. [25] reported the case of a patient who developed acute hepatitis B after infection with the subgenotype F1b, despite having undergone complete vaccination. Mutations in the S gene were not identified, mutations classically associated with escape of anti-HBs antibodies, such as sG145R, but the authors highlight the marked differences in the sequence of the S gene of the F genotype in relation to the other genotypes. The study by Limeres et al. [26] strengthens the evidence of the influence of the S gene variability of the F genotype, which we can extend to the H genotype, in the reduction of the binding capacity to anti-HBs antibodies. In that study, the authors evaluated the influence of the genetic variability of the F genotype S gene on the sensitivity of two widely used commercial kits for HBsAg research, and observed that the detection rates of this antigen produced by the subgenotypes F1b and F4 differed significantly from those obtained with antigens produced by the HBV genotype A.
Both genotypes have stop codon mutations in the envelope genes (S and preS). As we have used a next generation methodology, we were able to detect very low percentage of sequence variations in the patient samples. These mutations might affect the viral load found in each case as they may have severe impacts in the assembling of viable virus particles, particularly those stop codons found in the small S coding region. These mutations were particularly common among cases harboring genotype H that generally have very low viral loads. Some mutations found in the major hydrophilic region of HBsAg have also been identified especially in genotype H cases. Non silent mutations have been previously reported in occult hepatitis B infections (OBI) in Mexico and may explain the high number of such cases in this country [27].
Basal core promoter mutations have been reported more frequently subgenotypes F1b and F2 than in the other subgenotypes or genotype H. Genotypes F1b and F2 may be associated with a severe presentation of liver disease as opposed to a more benign course for subgenotypes F3 and F4 and genotype H. [28] Mutation in BCP has also been frequently found also in Brazilian F2 isolates [29].
Just as mutations in different regions of the HBV genome have been correlated with antiviral resistance, vaccine escape and hepatocellular carcinoma, coinfection and viral recombination events can also trigger greater virulence and worsen the clinical status of patients. Recombination is easy to detect in the process of assembling phylogenetic trees, however, the co-infection of two different genotypes can be mainly the result of artifacts during PCR amplification or by the wrong processing of sequence data, leading to the erroneous conclusion that there is a recombination. In 2000, Morozov et al. [30] claimed that to obtain reliable sequences from the viral co-infection event, it is advisable that the nucleotide sequence of the complete genome be made using a single PCR fragment. In the present study, there was also the characterization of a viral coinfection between the genotypes F1b and F3, made through the amplification of 3215bp of the HBV genome using primers P1 and P2 described by Gunther et al. [8].
The distribution of mutations in the HBV genome with clinical implications represents a challenge for diagnosis and different treatment strategies, and may even directly impact the long-term success of population vaccination programs [31]. Knowing the prevalence of clinically relevant mutations among different genotypes and subgenotypes facilitates the improvement of diagnostic procedures, immunization programs, therapeutic protocols, and disease prognosis [32]. These mutations can be analyzed by genome region and comparing the nucleotide sequence of the specific gene and its impact on the amino acid sequence.
As such, NGS has increasingly proven to be a powerful tool in medical diagnosis. Through this technique, it is possible to sequence the HBV genome and it is possible to identify, in addition to mixed genotypes and quasispecies of the virus, variations and polymorphisms that are important in the assessment of drug resistance.AbbreviationsBCP Basal core promoter Basic Local Alignment Search Tool base pair desoxyribonucleic acid guanine + cytosine Hepatitis B surface antigen Hepatitis B virus microliter minute sodium hydroxide polymerase chain reaction picomol pre core region pre S region second surface antigen gene Tris-acetate-EDTA Thermus aquaticus times Hepatitis B X gene
This work was partially supported by MCTI/CNPQ/Universal 14/2014 grant received by Dr. Rodrigo dos Santos Francisco.



