











Autoimmune hepatitis has a global occurrence, diverse clinical phenotype, and evolving treatment options. The goals of this report are to review the codified diagnostic criteria, spectrum of clinical presentations, proposed pathogenic mechanisms, conventional treatment strategies, and promising interventions. The literature published in English from 1980-2005 was reviewed and an updated current perspective provided. Autoimmune hepatitis affects all ages, may be asymptomatic, frequently has an acute onset, and can present as fulminant hepatitis. Perivenular (zone 3) necrosis is within the histological spectrum. Autoimmune hepatitis can recur or develop de novo after liver transplantation. CD4+ T-helper cells and natural killer T cells have been implicated in the pathogenesis, and molecular mimicry may break self-tolerance. DRB1*0301 and DRB1*0401 are the susceptibility alleles among white North Americans and northern Europeans, whereas diverse alleles of HLA DR4 have been associated with the disease in Japan, mainland China, and Mexico. DRB1*1301 is associated with autoimmune hepatitis in South American children, and it may predispose to an indigenous etiologic agent. Antibodies to soluble liver antigen/liver pancreas may have prognostic importance, and cyclosporine and mycophenolate mofetil must be assessed by clinical trial before incorporation into management algorithms. Sitespecific interventions are feasible, and they require a confident experimental animal model for evaluation. Variant syndromes lack diagnostic and therapeutic guidelines. In conclusion, autoimmune hepatitis must be considered in all patients with acute and chronic liver disease and those with allograft dysfunction after transplantation. New immunosuppressive agents and site-specific interventions promise to improve care.
Autoimmune hepatitis is an unresolving inflammation of the liver of unknown cause.1 It is characterized by interface hepatitis on histological examination, hypergammaglobulinemia, and autoantibodies. The sine qua non of the diagnosis is the presence of interface hepatitis on histological examination. Lymphocytic, often lymphoplasmacytic, inflammatory infiltrates extend from portal tracts into parenchymal tissue where they are associated with hepatocyte injury (Figure 1). Parenchymal inflammation may be limited to periportal areas (interface hepatitis), or it may involve the entire acinus (panacinar or lobular hepatitis) (Figure 2). Plasma cells can be seen within the infiltrate (Figure 3), but they are not specific or essential for the diagnosis. Plasma cells in groups or sheets in the portal tracts are present in 66% of patients, and this finding in conjunction with moderate-to-severe interface hepatitis and/or panacinar hepatitis has a diagnostic specificity of 81% and positive predictability of 68%.2



Autoimmune hepatitis has a global occurrence, and it has been described in African Americans, native Alaskans, Arabs, Asians, Europeans, Iranians, South Americans, and subcontinental Indians. Its incidence among white northern Europeans is 1.9 cases per 100,000 persons per year, and its point prevalence is 16.9 cases per 100,000 persons per year.3,4 In the United States, autoimmune hepatitis affects 100,000 to 200,000 persons, and it accounts for 5.9% of the liver transplantations in this country and 2.6% of the liver transplantations in Europe.3 The frequency of autoimmune hepatitis among patients with chronic liver disease in North America is between 11% and 23%. Its prevalence among Alaskan natives is 43 per 100,000 population and higher than that reported elsewhere.4
Race may affect disease severity as well as occurrence. Cirrhosis is present at accession more commonly in black North American patients with autoimmune hepatitis than in white North American patients (85% versus 38%), and hepatic synthetic function is decreased more frequently.7Alaskan natives have a higher frequency of acute icteric disease, asymptomatic illness, and advanced fibrosis at presentation than non-native counterparts.6 Japanese patients typically have mild, late onset disease that can respond to non-steroidal medication such as ursodeoxycholic acid.8 South American patients are younger than white North American counterparts, and they have more severe laboratory abnormalities.9 African, Asian and Arab patients have early age onset disease, and they have a higher frequency of cholestatic laboratory findings, greater occurrence of biliary changes on histological examination, and poorer initial response to standard therapy than other ethnic groups.10 These findings suggest that geographical location and ancestry affect occurrence and behavior of the disease. Interwoven into the natural history of the disease in each racial group and geographical region are cultural and socioeconomic factors that remain unsorted. Differences in the consequences of the liver disease must be correlated with delays in diagnosis or difficulties in accessing medical care that are region-specific.11
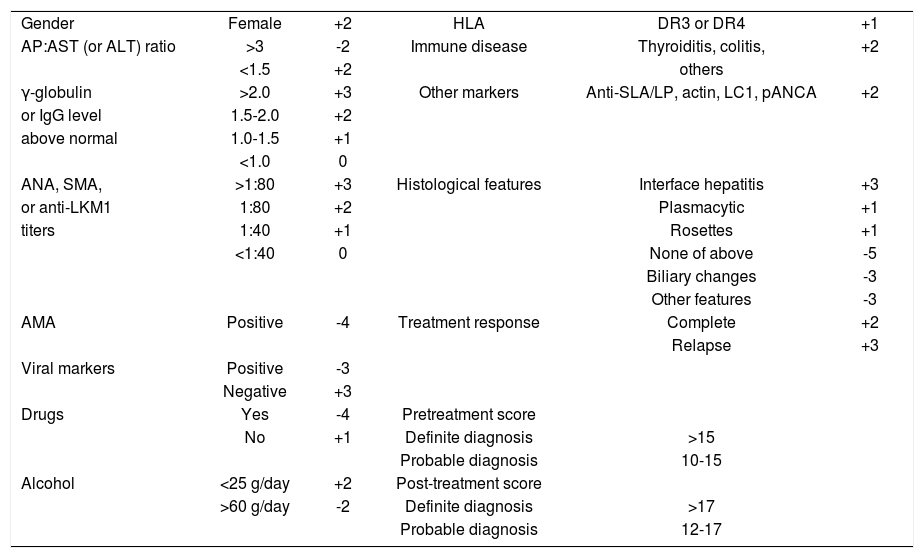
Diagnostic clinical criteriaThe clinical diagnosis of autoimmune hepatitis has been codified by an international panel, and these criteria must be applied to all patients12(Table I). An acute, even fulminant, presentation is recognized and important to diagnose quickly and treat promptly.13-16 The histological patterns that characterize acute onset autoimmune hepatitis are a panacinar hepatitis that resembles an acute viral or drug-induced hepatitis16,17(Figure 2) and a centrilobular or perivenular (Rappaport zone 3) hepatitis that resembles an acute toxic injury16,18,19(Figure 4).
International criteria for the diagnosis of autoimmune hepatitis.
| Diagnostic Criteria | |
|---|---|
| Definite AIH | Probable AIH |
| Normal α-1 AT phenotype | Partial α-1 AT deficiency |
| Normal ceruloplasmin level | Nondiagnostic ceruloplasmin/copper levels |
| Normal iron and ferritin levels | Nondiagnostic iron and/or ferritin changes |
| No active hepatitis A, B and/or C infection | No active hepatitis A, B and/or C infection |
| Daily alcohol <25 g/day | Daily alcohol <50 g/day |
| No recent hepatotoxic drugs | No recent hepatotoxic drugs |
| Predominant serum AST/ALT abnormality | Predominant serum AST/ALT abnormality |
| Globulin, γ-globulin or IgG level ≥1.5 times upper limit of normal ANA, SMA, or anti-LKM1 ≥1:80 in adults and ≥1:20 in children; no AMA | Hypergammaglobulinemia of any degree ANA, SMA or anti-LKM1≥1:40 in adults; other autoantibodies |
| Interface hepatitis, moderate to severe | Interface hepatitis, moderate to severe |
| No biliary lesions, granulomas or prominent changes suggestive of another disease | No biliary lesions, granulomas or prominent changes suggestive of another disease |
AIH = autoimmune hepatitis; α-1 AT = alpha-1 anti-trypsin; ANA = antinuclear antibodies; SMA = smooth muscle antibodies; anti-LKM1 = antibodies to liver/kidney microsome type 1; AMA = antimitochondrial antibodies; IgG = serum immunoglobulin G level.
 necrosis. Inflammation and hepatocyte drop out are present around a terminal hepatic venule. Mild diffuse inflammation and disorganization of the hepatic plate architecture are also present. Hematoxylin and eosin. Original magnification, x200.")
Transitions from a perivenular (Rappaport zone 3) hepatitis to an interface hepatitis have been demonstrated in successive biopsy specimens from patients with acute onset disease.19 These observations suggest that the perivenular (Rappaport zone 3) pattern of injury may be an early histological manifestation of autoimmune hepatitis that is unrecognized in specimens obtained later in the course. An exacerbation of a previously unrecognized chronic liver disease may also have an acute presentation, and it should be suspected by the presence of hypoalbuminemia, thrombocytopenia, ascites, esophageal varices, and/or bridging (septal) fibrosis or cirrhosis on histological examination.17,20
There are no disease-specific clinical or histological features. The diagnosis of autoimmune hepatitis requires the confident exclusion of other similar disorders including Wilson disease, genetic hemochromatosis, α-1 antitrypsin deficiency, chronic viral hepatitis, drug-related chronic liver disease, primary biliary cirrhosis (PBC), and primary sclerosing cholangitis (PSC)12(Table I). Minocycline is the drug that has been most commonly implicated as a cause of the syndrome.21 A cholestatic form of autoimmune hepatitis is not recognized, and the presence of pruritus or hyperpigmentation compels another diagnosis.1,12
Diagnostic scoring criteriaAn international scoring system for the objective diagnosis of the disease has been developed and validated retrospectively12,22,23(Table II). Multiple clinical, laboratory and histological features are graded, and a composite score is derived both before and after corticosteroid treatment. The scoring system was developed as a research tool to ensure comparability between patient populations in clinical trials. It is not a discriminative diagnostic index, and it should not be used to distinguish between classical syndromes of chronic liver disease or to deduce that common non-disease-specific clinical and laboratory features connote a mixed or hybrid pathological state.
International scoring system for diagnosis of autoimmune hepatitis.
| Gender | Female | +2 | HLA | DR3 or DR4 | +1 |
| AP:AST (or ALT) ratio | >3 | -2 | Immune disease | Thyroiditis, colitis, | +2 |
| <1.5 | +2 | others | |||
| γ-globulin | >2.0 | +3 | Other markers | Anti-SLA/LP, actin, LC1, pANCA | +2 |
| or IgG level | 1.5-2.0 | +2 | |||
| above normal | 1.0-1.5 | +1 | |||
| <1.0 | 0 | ||||
| ANA, SMA, | >1:80 | +3 | Histological features | Interface hepatitis | +3 |
| or anti-LKM1 | 1:80 | +2 | Plasmacytic | +1 | |
| titers | 1:40 | +1 | Rosettes | +1 | |
| <1:40 | 0 | None of above | -5 | ||
| Biliary changes | -3 | ||||
| Other features | -3 | ||||
| AMA | Positive | -4 | Treatment response | Complete | +2 |
| Relapse | +3 | ||||
| Viral markers | Positive | -3 | |||
| Negative | +3 | ||||
| Drugs | Yes | -4 | Pretreatment score | ||
| No | +1 | Definite diagnosis | >15 | ||
| Probable diagnosis | 10-15 | ||||
| Alcohol | <25 g/day | +2 | Post-treatment score | ||
| >60 g/day | -2 | Definite diagnosis | >17 | ||
| Probable diagnosis | 12-17 |
AP:AST (or ALT) ratio=ratio of alkaline phosphatase level to aspartate or alanine aminotransferase level; anti-SLA/LP=antibodies to soluble liver antigen/liver pancreas; anti- LC1=antibodies to liver cytosol type 1; pANCA=perinuclear anti-neutrophil cytoplasmic antibodies; IgG=immunoglobulin G; ANA=antinuclear antibodies; SMA=smooth muscle antibodies; anti-LKM1=antibodies to liver/kidney type 1; AMA=antimitochondrial antibodies; and HLA=human leukocyte antigen.
The virtues of the scoring system are that it quantifies the diagnosis, facilitates objective comparisons between patient populations, and accommodates individuals with atypical manifestations. Its drawbacks are its complexity, and its failure to consistently distinguish cholestatic syndromes from autoimmune hepatitis. The sensitivity of the scoring system for autoimmune hepatitis ranges from 97%-100%, and its specificity for excluding autoimmune hepatitis in patients with chronic hepatitis C ranges from 66%-92%. Its diagnostic specificity for excluding autoimmune hepatitis in cholestatic syndromes ranges from 45%-65%.12
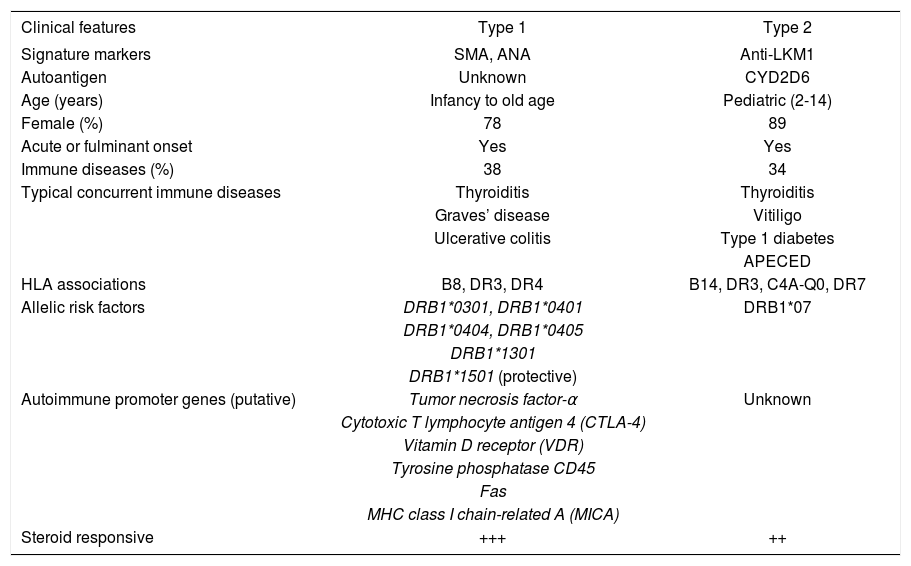
TypesThree types of autoimmune hepatitis have been proposed based on serological markers, but only two types have distinctive clinical phenotypes24(Table III). None has been ascribed a unique cause, individual management strategy or special behavior, and they have not been endorsed as separate entities by the International Autoimmune Hepatitis Group. The designations are used mainly as clinical descriptors.
Types of autoimmune hepatitis
| Clinical features | Type 1 | Type 2 |
|---|---|---|
| Signature markers | SMA, ANA | Anti-LKM1 |
| Autoantigen | Unknown | CYD2D6 |
| Age (years) | Infancy to old age | Pediatric (2-14) |
| Female (%) | 78 | 89 |
| Acute or fulminant onset | Yes | Yes |
| Immune diseases (%) | 38 | 34 |
| Typical concurrent immune diseases | Thyroiditis | Thyroiditis |
| Graves’ disease | Vitiligo | |
| Ulcerative colitis | Type 1 diabetes | |
| APECED | ||
| HLA associations | B8, DR3, DR4 | B14, DR3, C4A-Q0, DR7 |
| Allelic risk factors | DRB1*0301, DRB1*0401 | DRB1*07 |
| DRB1*0404, DRB1*0405 | ||
| DRB1*1301 | ||
| DRB1*1501 (protective) | ||
| Autoimmune promoter genes (putative) | Tumor necrosis factor-α | Unknown |
| Cytotoxic T lymphocyte antigen 4 (CTLA-4) | ||
| Vitamin D receptor (VDR) | ||
| Tyrosine phosphatase CD45 | ||
| Fas | ||
| MHC class I chain-related A (MICA) | ||
| Steroid responsive | +++ | ++ |
SMA = smooth muscle antibodies; ANA = antinuclear antibodies; anti-LKM1 = antibodies to liver/kidney microsome type 1; CYD2D6 = cytochrome 2D6; APECED = polyendocrinopathy-candidiasis-ectodermal dystrophy; MHC = major histocompatibility complex.
Type 1 autoimmune hepatitis is the most common form worldwide, constituting 80% of all cases, and it is characterized by the presence of antinuclear antibodies (ANA) and/or smooth muscle antibodies (SMA) (Table III). Seventy-eight percent of patients are female, and the female:male ratio is 3.5. Earlier reports that autoimmune hepatitis had a bimodal age distribution between ages 10 years and 30 years and between 40 years and 50 years were probably affected by referral patterns to tertiary medical centers.25 Current experiences suggest that autoimmune hepatitis occurs as commonly across all age ranges and that it may be under-diagnosed in the elderly.26,27 Forty-eight percent of patients are less than 40 years old, and the disease can affect infants.lin G level.
An abrupt onset of symptoms occurs in 40%, and a fulminant presentation is possible.16 Thirty-eight percent of individuals have concurrent immune diseases, especially autoimmune thyroiditis, synovitis, or ulcerative colitis, and 25% have cirrhosis already established at the time of presentation.28 Cholangiography is warranted in patients with ulcerative colitis to exclude PSC. Forty-one percent will have abnormal cholangiograms that support the diagnosis of PSC, and this finding may explain a refractory response to corticosteroid therapy.29 The high frequency of cirrhosis at presentation indicates that type 1 autoimmune hepatitis has an indolent, aggressive stage.
Thirty-four percent of patients with type 1 autoimmune hepatitis may be asymptomatic at initial consultation, and they are most commonly men with lower serum aminotransferase and immunoglobulin levels than symptomatic patients.31 Histological features are similar between asymptomatic and symptomatic patients, and both groups respond as well to corticosteroids. Most asymptomatic patients become symptomatic during follow-up, and differences between the asymptomatic and symptomatic state may reflect variations in disease activity and patient tolerance.
Type 2 autoimmune hepatitis is characterized by the presence of antibodies to liver/kidney microsome type 1 (anti-LKM1)31(Table III). This disease occurs mainly in children, but 20% of patients with type 2 disease in Europe are adults. Concurrent immune diseases are also common, especially insulin-dependent diabetes mellitus, vitiligo, and autoimmune thyroiditis. Organ specific autoantibodies are frequent, including antibodies to parietal cells, islets of Langerhans, and thyroid. As in type 1 disease, a fulminant presentation is possible and important to recognize early.14
Type 2 autoimmune hepatitis is the only form in which the target autoantigen has been identified. It is the cytochrome mono-oxygenase, CYP2D6, which is an important drug-metabolizing enzyme within the cytosol of the hepatocyte.32-34 The antigen has been sequenced, cloned, and mapped, and five antigenic sites located between peptides 193-212, 257-269, 321-351, 373-389, and 410-429 are recognized by anti-LKM1.35 The amino acid sequence spanning 193-212 of the CYP2D6 molecule is the target of anti-LKM1 in 93% of patients.
Homologies have been recognized between epitopes on the CYP2D6 molecule and the genome of the hepatitis C virus (HCV).34-37 The detection of anti-LKM1 in occasional patients with chronic hepatitis C in Europe (≤10%) may reflect this molecular mimicry and antibody crossreactivity. The hexameric amino acid sequence spanning 193-212 of the CYP2D6 molecule is homologous to the sequence spanning region 2985-2990 of the HCV genome and identical to the sequence spanning region 130-135 of the cytomegalovirus (CMV) genome.35 These homologies suggest that multiple exposures to viruses mimicking self may be a mechanism by which to break self-tolerance and induce type 2 autoimmune hepatitis. Antibodies to LKM1 are extremely rare in North American patients with chronic hepatitis C, and this rarity may reflect differences in the indigenous virus or the genetic susceptibility of the host.38,39 Studies in Germany40 and Italy41 have not found an association between structural changes within the viral genome and the presence of anti-LKM1. A host factor for anti-LKM1 expression has been implicated.40
Type 2 autoimmune hepatitis occurs in 15% of patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED).42 This syndrome consists of multiple endocrine organ failure, mucocutaneous candidiasis, and ectodermal dystrophy in various syndromic combinations that may include autoimmune hepatitis. APECED is caused by a single-gene mutation located on chromosome 21q22.3 which affects the generation of the autoimmune regulator (AIRE).43 AIRE is a transcription factor that is expressed in epithelial and dendritic cells within the thymus, and it regulates clonal deletion of autoreactive T cells (negative selection). APECED has an autosomal recessive pattern of inheritance, and it lacks HLA-DR associations and female predilection. The autoantigens associated with APECED are CYP1A2 and CYP2A6.
Type 3 autoimmune hepatitis is the least established form, and it is characterized by the presence of antibodies to soluble liver antigen/liver-pancreas (anti-SLA/LP).44,45 These antibodies are directed against a 50 kDa cytosolic protein46 which is a transfer RNA complex (tRNP(ser)sec) involved in the incorporation of selenocysteine into polypeptide chains.47 Patients with type 3 disease are mostly women (91%) with a mean age of 37 years (range, 17 to 67 years).44 Other autoantibodies, including ANA, SMA and anti-LKM1, can co-exist with anti-SLA/LP, and only 26% of patients have anti-SLA/LP as their sole serological finding.44 Patients with anti-SLA/LP are indistinguishable from patients with type 1 autoimmune hepatitis by clinical or laboratory features, HLA phenotype, or response to corticosteroids.48,49 The designation of a type 3 autoimmune hepatitis has been largely abandoned.
Pathogenic mechanismsThe pathogenic mechanisms of autoimmune hepatitis are unknown. The most popular hypotheses evoke a constellation of interactive factors that include a triggering agent, a genetic predisposition, and various determinants of autoantigen display, immunocyte activation, and effector cell expansion.50,51 Multiple triggering factors have been proposed, and they include infectious agents, drugs, and toxins. The multiplicity of etiologic agents that have been implicated in the pathogenesis of the disease suggests that the triggering epitope is a short amino acid sequence that is common in many antigens. There can be a long lag time between exposure to the trigger and onset of the disease, and the triggering factor is not needed for perpetuation of the disorder.
The CD4+ T-helper cell is the principal effector cell, and its activation and differentiation are the initial steps in the pathogenic pathway50,51(Figure 5). Natural killer T (NKT) cells are abundant in the liver, and they have also been implicated in the pathogenesis of the disease. NKT cells are produced in the bone marrow rather than the thymus, lack antigen-specific receptors, and produce interferon (IFN)-γ and tumor necrosis factor (TNF)-α. They are inhibited by cells with normal expression of the major histocompatibility complex (MHC) and by inhibitory receptors activated by glycolipid. Conversely, they target cells with aberrant MHC expression, defend against cells altered by viruses or cancer, and seem to promote hepatic regeneration.
 and tumor necrosis factor-α (TNF-α). The type 1 cytokine response promotes clonal expansion of liver infiltrating cytotoxic T lymphocytes (CD8 CTL). The type 2 cytokine response promotes the expansion of plasma cells and immunoglobulin G (IgG) production. The immunoglobulin complexes with normal membrane proteins on the hepatocyte surface, and natural killer T (NKT) cells target these complexes and cause cytolysis by an antibody-dependent cellmediated form of cyotoxicity. Type 1 and type 2 cytokine responses are counter-regulatory.")
Cytokine pathways of immunocyte differentiation. The principal effector is the CD4+ T helper cell, and its differentiation depends on the counter-regulatory effects of the interleukins (IL) and tumor necrosis factor-α (TNF-α). The type 1 cytokine response promotes clonal expansion of liver infiltrating cytotoxic T lymphocytes (CD8 CTL). The type 2 cytokine response promotes the expansion of plasma cells and immunoglobulin G (IgG) production. The immunoglobulin complexes with normal membrane proteins on the hepatocyte surface, and natural killer T (NKT) cells target these complexes and cause cytolysis by an antibody-dependent cellmediated form of cyotoxicity. Type 1 and type 2 cytokine responses are counter-regulatory.
The immunoregulatory cytokines orchestrate immunocyte differentiation through cross-regulatory actions and result in cellular and humoral mechanisms of liver cell injury50-52(Figure 5). Interleukin (IL)-2, IFN-γ, and TNF-α constitute the type 1 (Th1) cytokine response which regulates cellular immune mechanisms by facilitating clonal expansion of cytotoxic T lymphocytes. Interleukin-4, IL- 5, IL-6, IL-8, IL-10 and IL-13 constitute the type 2 (Th2) cytokine response which influences the humoral immune response by activating B cells and stimulating autoantibody production. The type 1 cytokine response favors liver injury by expanding sensitized tissue-infiltrating cytotoxic T cells (cellular cytotoxicity), and the type 2 cytokine response favors liver cell injury by generating immunoglobulin complexes on the hepatocyte surface that are targeted by NKT cells (antibody-dependent cellmediated cytotoxicity). The type 2 cytokine response also has anti-inflammatory effects that counter the type 1 cytokine actions.
Molecular mimicry of a foreign antigen and a self-antigen is the most common explanation for the loss of selftolerance, but this mechanism has not been established in human disease.51 Cross-reacting autoantibodies between foreign and self antigens have been described, but crossreacting immunocytes have been more difficult to demonstrate. Recently, a murine model of type 2 autoimmune hepatitis based on DNA immunization against self-antigens has supported this possibility.53 Most immunized mice developed peak serum alanine aminotransferase abnormalities 4 and 7 months after the last of three plasmid injections that contained the antigenic regions of human CYP2D6 and human formiminotransferase cyclodeaminase, the target antigen of antibodies to liver cytosol type 1 (anti-LC1). Affected mice expressed anti-LKM1 and anti-LC1, but they also had cytotoxic T lymphocytes within the liver that were sensitized against the antigens in the plasmid constructs. This murine model indicated that DNA immunization against human autoantigens could break self-tolerance and cause liver injury by molecular mimicry between foreign and self-antigens involving cross-reacting humoral and cellular responses.
HLA AssociationsGenetic factors affect the occurrence, clinical expression and treatment outcome of type 1 autoimmune hepatitis. HLA DR3 is the main susceptibility factor in white northern European and North American patients, and HLA DR4 is a secondary but independent risk factor54(Table III). Eighty-five percent of white patients from these regions have HLA DR3, DR4 or DR3-DR4. Different geographical regions and ethnic groups have different susceptibility factors. HLA DR3 occurs rarely in the Japanese population, and HLA DR4 is the principal risk factor for autoimmune hepatitis in this ethnic group.55 HLA DR4 is also the principal susceptibility factor for autoimmune hepatitis in mainland China.56 In contrast, HLA DR3, but not HLA DR4, is the susceptibility factor in Italy,57 and HLA DR13 is associated with childhood autoimmune hepatitis in South America.9,58-60
Patients with HLA DR4 in North America are older and more commonly female than patients with HLA DR3.61 They also have higher serum levels of γ-globulin and immunoglobulin G (IgG), higher titers of ANA, and a greater frequency of concurrent immune diseases. Patients with HLA DR4 respond more readily to corticosteroid therapy than those with HLA DR3 by entering remission more commonly and failing treatment less often. The bases for these effects on clinical phenotype and outcome are unknown, but they may relate to the diversity of alleles associated with each susceptibility factor.
HLA DR3 is associated with only 2 alleles that might affect autoreactivity, and DRB1*0301 is the only allele common in the United States.62 In contrast, HLA DR4 is associated with 26 alleles that may affect autoreactivity, of which 10 are common in the United States. HLA DR4- positive patients have a greater diversity of susceptibility alleles, and they can present a wider spectrum of antigens to immunocytes than patients with HLA DR3. This diversity of antigenic presentation may in turn enhance the spectrum of clinical manifestations and the frequency of concurrent immune diseases.
The genetic associations of type 2 autoimmune hepatitis are not well defined because the disease is infrequent in some geographical regions and type 1 autoimmune hepatitis is the predominant form in all experiences. Patients with type 2 autoimmune hepatitis from Germany have DRB1*03 and DRB1*04 less commonly and DRB1*07 more frequently than white North American patients with type 1 autoimmune hepatitis and normal control subjects63(Table III).DRB1*07 has also been associated with the disease in Brazil,64 whereas HLA B14, HLA DR3 and C4A-QO have been incriminated as genetic risk factors in northern Europe.65 Recent studies have suggested that the expression of anti-LKM1 may be associated with DRB1*07 regardless of the autoimmune or viral basis of the liver disease.66 Host-specific genetic factors in addition to disease-specific etiologic agents probably contribute to the production of anti-LKM1 and influence its occurrence in different geographical areas and racial groups. Unlike type 1 autoimmune hepatitis, the HLA phenotype of type 2 autoimmune hepatitis has not yet been ascribed a clinical relevance.
Allelic associationsHigh resolution DNA-based techniques have indicated that the alleles associated with susceptibility, clinical expression and outcome in white northern European and North American patients with type 1 autoimmune hepatitis are DRB1*0301 and DRB1*0401.67 These findings implicate the DRB1 locus as the principal susceptibility region of the MHC. Patients with DRB1*0301 are younger than patients with DRB1*0401, and they fail corticosteroid therapy more often, die of liver failure or require liver transplantation more commonly, and have a significantly greater frequency of an adverse treatment outcome than patients with DRB1*0401.68
The risk of type 1 autoimmune hepatitis may relate to amino acid sequences in the antigen binding groove of the class II MHC molecule, and multiple alleles may encode the same or similar sequence69,70(Figure 6). The critical shared motif in white North Americans and northern Europeans with type 1 autoimmune hepatitis is a sixamino- acid sequence represented by the code, LLEQKR.67,71 This sequence is located between positions 67 and 72 of the DRβ polypeptide chain of the class II MHC molecule, and lysine (K) in position 71 is the critical determinant of susceptibility.
 is the critical sequence encoded by the susceptibility alleles. DRβ*0301 and DRB1*0401 encode the identical sequence of LLEQKR (insert) where lysine (K) is the critical determinant at position DRβ71. DRB1*0404 and DRB1*0405, which are the susceptibility alleles in Mexican, Japanese, mainland Chinese and Argentine adults, encode a similar sequence except for an arginine (R) at the DRβ71 position. DRB1*1501, which protects against type 1 autoimmune hepatitis in white North Americans and northern Europeans, encodes an isoleucine (I) for leucine (L) at position DRβ67 and an alanine (A) for lysine (K) at position DRβ71. DRB1*1301, which is associated with type 1 autoimmune hepatitis in Argentine children and Brazilian patients, encodes ILEDER at positions DRβ67-72 where glutamic acid (E), aspartic acid (D), and glutamic acid (E) are at positions DRβ69, 70 and 71, respectively. The structural and electrostatic properties of the antigen binding groove determine the antigens that can be presented.")
Key susceptibility sequence within the antigen binding groove of the class II MHC molecule. The six amino acid sequence between positions 67 and 72 on the DRβ chain of the class II MHC molecule (insert) is the critical sequence encoded by the susceptibility alleles. DRβ*0301 and DRB1*0401 encode the identical sequence of LLEQKR (insert) where lysine (K) is the critical determinant at position DRβ71. DRB1*0404 and DRB1*0405, which are the susceptibility alleles in Mexican, Japanese, mainland Chinese and Argentine adults, encode a similar sequence except for an arginine (R) at the DRβ71 position. DRB1*1501, which protects against type 1 autoimmune hepatitis in white North Americans and northern Europeans, encodes an isoleucine (I) for leucine (L) at position DRβ67 and an alanine (A) for lysine (K) at position DRβ71. DRB1*1301, which is associated with type 1 autoimmune hepatitis in Argentine children and Brazilian patients, encodes ILEDER at positions DRβ67-72 where glutamic acid (E), aspartic acid (D), and glutamic acid (E) are at positions DRβ69, 70 and 71, respectively. The structural and electrostatic properties of the antigen binding groove determine the antigens that can be presented.
DRB1*0301 and DRB1*0401 encode identical amino acid sequences in the DRβ 67-72 region, and they affect susceptibility similarly.67DRB1*0404 and DRB1*0405 are the susceptibility alleles in Mexican,72 Japanese,55 mainland Chinese56 and Argentine adults,58,59 and they encode a similar sequence except for an arginine (R) for lysine (K) at the DRβ 71 position. Arginine is a positively charged amino acid that is structurally similar to lysine, and its substitution for lysine would not greatly alter the antigen binding properties of the class II MHC molecule.
In contrast, DRB1*1501 protects against type 1 autoimmune hepatitis in white North Americans and northern Europeans, and this allele encodes an isoleucine (I) for leucine (L) at position DRβ 67 and an alanine (A) for lysine (K) at position DRβ 7167,71(Figure 6). Alanine is a neutral, nonpolar amino acid whose substitution for lysine would greatly affect antigen presentation and immunocyte activation. As in other autoimmune diseases such as type 1 diabetes mellitus, the substitution of a single amino acid at a critical location in the antigen binding groove of the class II MHC molecule may affect disease occurrence. By understanding the requirements for optimal autoantigen presentation, it is possible to predict the ideal antigenic peptide. This ideal peptide must have a negatively charged residue, either aspartic acid or glutamic acid, at position P4 from the N terminus to complement the positively charged residue at position DRβ 71 (either lysine or arginine).70
DRB1*1301 is associated with type 1 autoimmune hepatitis in Argentine children and Brazilian patients, and it encodes ILEDER at positions DRβ 67-72.70,73 Glutamic acid (E), aspartic acid (D), and glutamic acid (E) are at positions DRβ 69, 70 and 71, respectively, in the class II MHC molecule. These critically located but negatively charged amino acid residues create a different antigenpresenting milieu within the class II MHC molecule than that encoded by the DRB1*0301 and DRB1*04 alleles. These findings have generated a “molecular footprint hypothesis” of pathogenesis which holds that susceptibility to type 1 autoimmune hepatitis in different regions and racial groups relates to indigenous factors or agents favored by certain genetic phenotypes.73
In South America, DRB1*1301 is associated with protracted hepatitis A virus infection,74 and individuals with this allele may be selected from their environment to have prolonged exposure to viral and hepatic antigens that favor the development of autoimmune hepatitis.75 The individual susceptibility allele in a geographic region may be a “footprint” by which to track the cause of the disease.
Autoimmune promotersGenetic autoimmune promoters inside and outside the MHC may also affect the occurrence of autoimmune hepatitis either in synergy with the principal susceptibility alleles (epistasis) or in lieu of them73(Table III). Polymorphisms of the tumor necrosis factor-α gene (TNFA*2)76,77 and the cytotoxic T lymphocyte antigen 4 gene (CTLA- 4)78,79 have been associated with increased immune reactivity and disease severity in type 1 autoimmune hepatitis in white North American and northern European patients. These promoters are host-related and not disease-specific. Constellations of them in varying combinations may affect the occurrence, clinical phenotype, and outcome of autoimmune hepatitis. Their occurrence and impact may also vary by geographical region and ethnic group.80
Other genetic promoters that have been implicated in the pathogenesis of autoimmune hepatitis include polymorphisms of the vitamin D receptor (VDR) gene,81 point mutation of the tyrosine phosphatase CD45 gene,82 polymorphisms of the Fas gene (tumor necrosis factor receptor super family-6 or TNFRSF6),83 and polymorphisms of the MHC class I chain-related A gene (MICA)84(Table III). Cytokine imbalances perhaps related to genetic polymorphisms that control cytokine production and receptor function are undoubtedly important in affecting the cascade of immune-mediated interactions resulting in hepatocyte injury. In this context, transforming growth factor-β (TGF-β) has recently been implicated as an important protective mechanism against autoimmune hepatitis by suppressing infiltration of the liver with autoreactive T cells.85
Gender effectsThe female predisposition for autoimmune hepatitis and autoimmune disease in general is unexplained.86 HLA DR4-positive women with type 1 autoimmune hepatitis have a greater variety of HLA DR4 alleles associated with their disease than HLA DR4-positive men.62 Women may thereby have a greater facility to be sensitized to self or foreign antigens than men. They may also be exposed to unique antigens, and/or they may respond to common antigens against which men do not react.
Triggering factors unique to the female state may include drugs, toxins, infections, and environmental antigens. Pregnancy is a unique state of women and protracted exposures to fetal cells or antigens in the maternal circulation may stimulate autoreactivity through microchimerism. The diversity and strength of the antigenic stimulation in women may in turn be modulated by the repertoire of HLA DR4 alleles and various hormonal factors.
Estrogen levels do modulate immune reactivity, and they may be contributory to the autoimmune propensity in women.87 High estrogen levels favor a type 2 cytokine response which drives activated immunocytes towards antibody production and an anti-inflammatory effect (Figure 5). Normal or low estrogen levels promote a type 1 cytokine response which drives the clonal expansion of tissue infiltrating cytotoxic T cells and causes liver damage (Figure 5).
The female propensity for autoimmune hepatitis is apparent among children and among pre-and post-menopausal adults.26,88 The presence or absence of estrogen, therefore, is an insufficient explanation for the risk of disease. Interactions between growth hormone, prolactin, testosterone and estrogen may constitute a changing hormonal milieu that affects immune responsiveness differently at various ages and favors certain antigens during different stages of maturation. The female gender may be the critical determinant affecting the hormonal blend of the interactive network at each age.
The importance of hormonal effects on the pathogenic mechanisms of autoimmune hepatitis is evident during pregnancy. Autoimmune hepatitis commonly improves during pregnancy, possibly because high estrogen levels promote a switch from type 1 cytokine actions which are cytotoxic to type 2 cytokine actions which are anti-inflammatory. Autoimmune hepatitis may then worsen after delivery, possibly because low estrogen levels promote a switch back to the type 1 cytokine actions which are cytotoxic.89 The role of other hormonal interactions during and after pregnancy in modulating this effect is unknown.
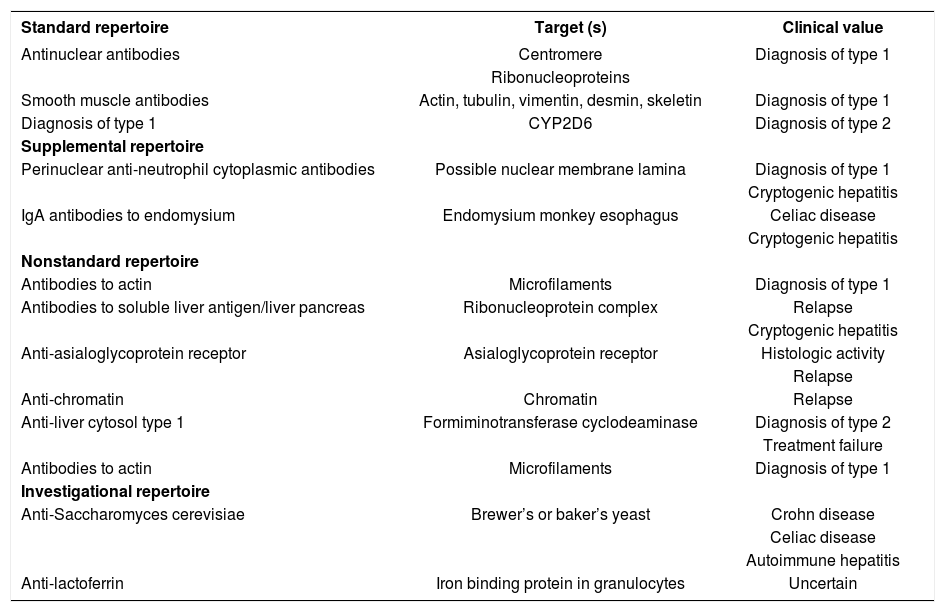
AutoantibodiesANA, SMA, and anti-LKM1 constitute the standard repertoire of autoantibodies that are assessed in autoimmune hepatitis1,12,90-92(Table IV). Antibodies to soluble liver antigen/liver pancreas (anti-SLA/LP), neutrophil cytoplasm (pANCA), endomysium, and tissue transglutaminase are ancillary markers that are useful in evaluating patients who are seronegative for the standard battery.92 Other autoantibodies that have been described in autoimmune hepatitis which are either not generally available, investigational in nature, or of limited clinical value are antibodies to asialoglycoprotein receptor (anti-ASGPR),93 actin,94 chromatin,95 liver cytosol type 1,96 double-stranded DNA,97 histones,98Saccharomyces cerevisiae,99-100 and lactoferrin101(Table IV).
Autoantibodies associated with autoimmune hepatitis
| Standard repertoire | Target (s) | Clinical value |
|---|---|---|
| Antinuclear antibodies | Centromere | Diagnosis of type 1 |
| Ribonucleoproteins | ||
| Smooth muscle antibodies | Actin, tubulin, vimentin, desmin, skeletin | Diagnosis of type 1 |
| Diagnosis of type 1 | CYP2D6 | Diagnosis of type 2 |
| Supplemental repertoire | ||
| Perinuclear anti-neutrophil cytoplasmic antibodies | Possible nuclear membrane lamina | Diagnosis of type 1 |
| Cryptogenic hepatitis | ||
| IgA antibodies to endomysium | Endomysium monkey esophagus | Celiac disease |
| Cryptogenic hepatitis | ||
| Nonstandard repertoire | ||
| Antibodies to actin | Microfilaments | Diagnosis of type 1 |
| Antibodies to soluble liver antigen/liver pancreas | Ribonucleoprotein complex | Relapse |
| Cryptogenic hepatitis | ||
| Anti-asialoglycoprotein receptor | Asialoglycoprotein receptor | Histologic activity |
| Relapse | ||
| Anti-chromatin | Chromatin | Relapse |
| Anti-liver cytosol type 1 | Formiminotransferase cyclodeaminase | Diagnosis of type 2 |
| Treatment failure | ||
| Antibodies to actin | Microfilaments | Diagnosis of type 1 |
| Investigational repertoire | ||
| Anti-Saccharomyces cerevisiae | Brewer’s or baker’s yeast | Crohn disease |
| Celiac disease | ||
| Autoimmune hepatitis | ||
| Anti-lactoferrin | Iron binding protein in granulocytes | Uncertain |
New autoantibodies continue to be characterized in the hope that they will reveal critical pathogenic mechanisms or have prognostic value. Antibodies to SLA/LP are present in 26% of patients with autoimmune hepatitis who are otherwise seronegative,44,45 and they are useful in reclassifying cryptogenic chronic hepatitis as autoimmune hepatitis.102 These autoantibodies also identify individuals who have more severe disease than seronegative counterparts and who invariably relapse after corticosteroid withdrawal.103-105 Since anti-SLA/LP are closely associated with HLA DR3, they may be surrogate markers of a genetic propensity for relapse or refractory disease.103,105 Analysis of the immunoprecipitated RNAs extracted from HeLa cell extracts is the most powerful, sensitive and specific method to detect anti-tRNP(ser)sec/SLA/LP autoantibodies, but an enzyme-linked immunosorbent assay (ELISA) is available as a commercial kit and performance parameters between the methods are comparable.106
Perinuclear anti-neutrophil cytoplasmic antibodies (pANCA) and immunoglobulin A (IgA) antibodies to endomysium (EMA) and tissue transglutaminase (tTG) are ancillary markers of autoimmune hepatitis that are available in the clinical laboratory91,92(Table IV). pANCA are found with great frequency (50%-92%) and in high titer in type 1 autoimmune hepatitis, and they can be the sole serological markers of this disease.107,108 IgA EMA have a sensitivity of 94% and specificity of 99% for celiac disease,109,110 and they are less likely to be falsely positive in chronic hepatitis than IgA antibodies to tTG.100,111-113 Serological screening for celiac disease is important in patients with autoimmune hepatitis and in patients with chronic undifferentiated liver disorders since celiac disease can occur coincidentally with autoimmune liver disease114,115 or cause liver dysfunction that may improve with gluten restriction.116-119
The autoantibodies that are still investigational and that have promise as prognostic indices include anti-ASGPR and anti-actin (Table IV). The presence of anti-ASGPR correlates with histological activity and the propensity to relapse after corticosteroid withdrawal.93,120 Continuation of treatment until disappearance of anti- ASGPR has been associated with a sustained remission. Antibodies to actin identify patients with a higher frequency of treatment failure and death from liver failure or requirement for liver transplantation than seronegative patients, but they are restricted to those individuals with SMA.94
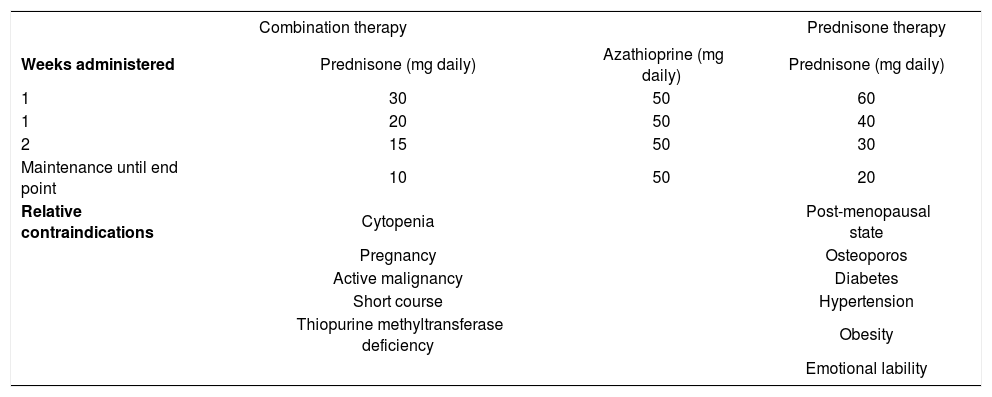
Conventional treatment schedulesThe preferred treatment schedule for all forms of autoimmune hepatitis in adults is prednisone in combination with azathioprine121-123(Table V). Prednisone alone in higher dose is as effective as the combination regimen, but it is associated with a higher frequency of drug-related side effects (44% versus 10%). The preferred treatment schedule in children is prednisone alone in a dose of 2 mg/kg daily (up to 60 mg daily).1 Azathioprine or 6-mercaptopurine can also be introduced as a corticosteroid- sparing measure. Tapering schedules or alternate day corticosteroid regimens are commonly used in children to reduce deleterious effects on linear growth, bone development, and physical appearance.
Conventional treatment regimens.
| Combination therapy | Prednisone therapy | ||
|---|---|---|---|
| Weeks administered | Prednisone (mg daily) | Azathioprine (mg daily) | Prednisone (mg daily) |
| 1 | 30 | 50 | 60 |
| 1 | 20 | 50 | 40 |
| 2 | 15 | 50 | 30 |
| Maintenance until end point | 10 | 50 | 20 |
| Relative contraindications | Cytopenia | Post-menopausal state | |
| Pregnancy | Osteoporos | ||
| Active malignancy | Diabetes | ||
| Short course | Hypertension | ||
| Thiopurine methyltransferase deficiency | Obesity | ||
| Emotional lability | |||
Therapy is continued until remission, treatment failure, incomplete response, or drug toxicity.122,123Remission implies the absence of symptoms, resolution of all laboratory indices of liver inflammation, and histological improvement to normal or minimal inflammatory activity. A serum aspartate aminotransferase level of less than twice the upper limit of normal is an acceptable laboratory end point if the histological examination confirms the absence of interface hepatitis. Treatment failure connotes deterioration during therapy, and it is characterized by worsening of the serum aspartate aminotransferase or bilirubin level by at least 67% of previous values, progressive histological activity, or onset of ascites or encephalopathy. Incomplete response connotes improvement that is insufficient to satisfy remission criteria after 3 years of continuous treatment, and drug toxicity implies severe intolerance of the medication. Histological improvement lags behind clinical and laboratory improvement by 3-8 months, and liver tissue examination prior to drug withdrawal is the only means of ensuring inactive disease.124,125
Treatment outcomesSixty-five percent of treated patients satisfy remission criteria within 18 months, and 80% achieve this result within 3 years.122,123 The average duration of treatment until remission is 22 months,28 and the 10-year life expectancies of treated patients with and without cirrhosis at accession are 89% and 90%, respectively.126 These survivals are comparable to those of an age-and sex-matched normal population from the same geographical region. Patients with cirrhosis respond as well to treatment as patients without cirrhosis, and they should be treated similarly with the same expectation of success.126,127 Twenty-one percent of individuals who enter remission sustain this result long term after drug withdrawal (median 7interval of follow-up, 76 months), and an effort should be made to discontinue initial therapy in all patients with inactive disease.128 Thirteen percent develop side effects that justify premature discontinuation of medication (drug toxicity); 9% deteriorate despite compliance with therapy (treatment failure); and 13% improve but not to a degree to satisfy criteria for remission (incomplete response).122,123
Corticosteroid therapy may also reduce hepatic fibrosis.129-132 Fibrosis scores improved in 56% of patients followed for 55+9 months, and they did not progress in 33% of patients followed for 62+14 months.132 Histological activity indices decreased concurrently, and patients in whom the histological activity indices improved had a higher frequency of improvement in the fibrosis scores (80% versus 25%, p=0.002).132,133 These findings suggest that improvement in hepatic fibrosis occurs in conjunction with reductions in liver inflammation and that corticosteroid therapy facilitates the disappearance of fibrosis by suppressing inflammatory activity. Small case studies have also suggested that cirrhosis can disappear during treatment, but this possibility must await confirmation by assays more reliably reflective of cirrhosis than conventional needle biopsy of the liver.129,132
Treatment of suboptimal responsesTreatment failure is managed by administering high dose prednisone alone (60 mg daily) or prednisone (30 mg daily) in conjunction with azathioprine (150 mg daily).122,123 Doses of prednisone and azathioprine are then reduced by 10 mg and 50 mg, respectively, for each month of laboratory improvement until conventional maintenance levels of drug are achieved. Seventy-five percent of patients treated in this fashion enter clinical and laboratory remission, but only 20% have histological resolution. These patients remain at risk for progressive liver disease and drug toxicity.
Drug toxicity requires premature dose reduction or discontinuation of the offending drug and continued use of the other tolerated medication in adjusted dose.122,123 Corticosteroid- related side effects are the most common causes for drug withdrawal, and they include intolerable cosmetic changes or obesity (47%), osteoporosis with vertebral compression (27%), brittle diabetes (20%), and peptic ulceration (6%).134 Azathioprine can be administered as a corticosteroid-sparing agent with doses increased to 2 mg/kg daily. The emergence of a cholestatic hepatitis, pancreatitis, rash, progressive cytopenia, or gastrointestinal upset indicates azathioprine toxicity and the need for its withdrawal. The dose of prednisone is then adjusted to suppress disease activity.
An incomplete response is declared after 3 years of conventional therapy without remission.122,123 Patients improve but not to a degree to satisfy remission criteria, and they are at risk for drug-related side effects associated with standard doses of prednisone. Low dose prednisone or long-term maintenance therapy with azathioprine (2 mg/kg daily) is a treatment option.
Relapse after remission and drug withdrawalRelapse occurs in 20%-86% of patients depending on the criteria for remission prior to drug withdrawal.135-137 Diminished stamina, arthralgias, and increase in the serum aspartate aminotransferase level to more than threefold normal characterize this occurrence. Re-treatment with the original regimen typically induces another remission, but relapse recurs in 79% within 6 months after drug withdrawal.135 With each re-treatment and relapse, the frequency of drug-related side effects increases, and this risk outweighs the low probability of a sustained remission with repeated conventional treatments.138 Alternative therapies with either low dose prednisone or azathioprine are warranted after the second relapse.
The low dose prednisone regimen requires induction of clinical and laboratory remission on standard therapy and then reduction in the dose of prednisone by 2.5 mg each month of clinical and laboratory stability.139 The lowest dose that prevents symptoms and keeps serum aspartate aminotransferase levels below three-fold normal is maintained. Eighty-seven percent of patients can be managed on prednisone, 10 mg daily or less (median dose, 7.5 mg daily). Side effects associated with earlier conventional treatments improve or disappear in 85%; new side effects do not develop; and survival is unaffected.
Maintenance therapy with azathioprine also requires induction of clinical and laboratory remission by conventional treatments.140,141 The corticosteroid component is then withdrawn, and the dose of azathioprine is increased to 2 mg per kg daily and maintained indefinitely. Eightyseven percent of adult patients managed in this fashion remain in remission during a median observation interval of 67 months. Follow-up liver biopsy assessments disclose inactive or minimal histological disease in 94%; corticosteroid-related side effects improve or disappear in most patients; and the drug is generally well-tolerated. The most common side effects are withdrawal arthralgias (63%), lymphopenia (57%), and myelosuppression (7%). Neoplasms involving diverse cell types occur in 8%.
Relapse does not preclude permanent discontinuation of medication late in the course of the disease.128,138 Twenty- eight percent of patients who relapse and are re-treated develop inactive disease and can be withdrawn from medication. The probability of a sustained remission after initial or subsequent therapy is 47% during 10 years of follow-up. Conventional re-treatment schedules are able to induce a sustained remission more commonly than long-term maintenance schedules (59% versus 12%, p=0.00002), but all management schedules should be withdrawn periodically to assess this outcome.
Liver transplantationLiver transplantation is an effective treatment for the decompensated patient. Patient and graft survival after liver transplantation ranges from 83% to 92%, and the actuarial 10-year survival after transplantation is 75%.5,142 Recurrence is recognized in at least 17% of patients after 5+1 years, especially in individuals receiving inadequate immunosuppression.143 Adjustments in the immunosuppressive regimen are usually able to suppress recurrent disease, but rarely cirrhosis or graft failure occurs.144 Patients transplanted for autoimmune hepatitis may also have a greater frequency of acute and chronic rejection than patients transplanted for non-autoimmune conditions.145,146 These potential consequences have tempered efforts to rapidly withdraw corticosteroids after the procedure.
Autoimmune hepatitis can develop de novo in children and adult recipients who undergo transplantation for nonautoimmune liver disease.147-152 Children seem to have a predilection for the syndrome; immunosuppression with cyclosporine is a common feature; and treatment with prednisone and azathioprine is typically effective. De novo autoimmune hepatitis is rare, occurring in 3%-5% of allografts, and it can result in graft loss if not treated with corticosteroids.153 In children, de novo disease may reflect defective negative selection of autoreactive cells by the thymus, generation of promiscuous lymphocytes by excessive antigenic exposure, and/or impaired apoptosis of autoreactive cells by cyclosporine or tacrolimus. In adult patients, these same mechanisms are pertinent except for thymic dysfunction.154,155
Emerging drug therapiesThere is no shortage of new drugs that have been proposed for autoimmune hepatitis156,157(Table VI). Many have emerged from the transplantation arena, but none has been rigorously evaluated or incorporated into a conventional management algorithm. Of the drugs that promise greater blanket immunosuppression than prednisone or azathioprine, cyclosporine and mycophenolate mofetil have shown the most promise. Controlled clinical trials are sorely needed to establish their efficacy.
Evolving Drug Therapies and Site-specific Interventions.
| New drug regimens | Site-specific Interventions | ||
|---|---|---|---|
| Agent | Possible Uses | Intervention | Mechanism(s) |
| Cyclosporine158-165 (5-6 mg/kg daily) | Treatment failure Steroid intolerance First-line therapy | Blocking synthetic peptides180 | Blocks 1st co-stimulatory signal |
| Mycophenolate mofetil167,168 (2 g daily) | Azathioprine intolerance Steroid withdrawal | Soluble cytotoxic T lymphocyte antigen-4182 | Blocks 2nd co-stimulatory signal |
| 6-mercaptopurine173 (1.5 mg/kg daily) | Azathioprine intolerance Treatment failure | Oral tolerance regimens183 | Dose dependent suppression immune response |
| Tacrolimus170,171 (4 mg twice daily) | Treatment failure Steroid intolerance | T cell vaccination184 | Clonal depletion of activated cytotoxic T lymphocytes |
| Budesonide172 (3 mg twice daily) | Front-line therapy Osteopenia | Cytokine manipulations186,187 | Recombinant cytokines Monoclonal antibodies |
| Ursodeoxycholic acid175 (13-15 mg/kg daily) | Mild disease in Japanese | Gene therapy188 | Limit fibrosis Promote repair Counter cytokines |
| Methotrexate176 | Treatment failure | ||
| Cyclophosphamide177 | Steroid intolerance | ||
| Deflazacort174 (7.5 mg for each 5 mg prednisone) | Steroid sparing | ||
Cyclosporine has been used successfully as salvage therapy in patients who have failed conventional corticosteroid treatment or been intolerant of the medication.158-165 It has also been used as first-line therapy in children and adults.163- 165 The medication binds cyclophilin and inhibits the phosphatase activity of calcineurin. As a calcineurin inhibitor, it impairs transcription of IL-2, cell cycle progression, and expansion of cytotoxic T lymphocytes. Side effects include renal insufficiency, hypertension, and malignancy.
Mycophenolate mofetil is an ester prodrug hydrolyzed by liver esterases to produce the active metabolite, mycophenolic acid, which in turn acts as a non-competitive, reversible inhibitor of inosine monophosphate dehydrogenase.166 Inosine monophosphate dehydrogenase is the ratelimiting enzyme for de novo synthesis of purines, and by inhibiting its action, mycophenolate mofetil selectively prevents the proliferative responses of T and B cells to mitogens or antigens. Mycophenolate mofetil is a purine antagonist like azathioprine, and it has a low frequency of side effects (mainly leukopenia) and independence from the thiopurine methyltransferase pathway of catabolism.
Two small uncontrolled clinical experiences have shown improvement in the laboratory indices of liver inflammation after the administration of mycophenolate mofetil, 1 gram twice daily, to patients unsuccessfully treated with azathioprine or intolerant of this drug.167,168 The prospect of replacing corticosteroids with mycophenolate mofetil has also stimulated its empiric use. Other preliminary experiences have failed to demonstrate a potent salvage effect or consistent corticosteroid-sparing action, and the proper role and target population for this drug remain uncertain.169
Tacrolimus (4 mg twice daily),170,171 budesonide (3 mg thrice daily),172 6-mercaptopurine (1.5 mg/kg daily),173 deflazacort (7.5 mg for each 5 mg prednisone dose daily),174 ursodeoxycholic acid (13-15 mg/kg daily),175 methotrexate,176 and cyclophospamide177 have all been used with anecdotal success in treating corticosteroid resistant or intolerant patients. Only ursodeoxycholic acid has been evaluated by randomized controlled clinical trial as a salvage therapy, and it is the one negative experience.178 Budesonide is currently undergoing clinical trial as frontline therapy after earlier studies had demonstrated its limitations as a salvage treatment.179
Emerging site-specific interventionsSite-specific interventions are designed to target key steps in the pathogenic pathway and thereby control the disease without inducing blanket immune suppression.157 These therapies are at theoretical or preliminary stages of development, and they await full clarification of the molecular mechanisms of the disease and the availability of suitable animal models to assess their feasibility. The various intracellular signaling pathways affecting immunocyte activation and proliferation are prime targets for these strategies.
Competitive inhibition of autoantigen presentation has already been applied in rheumatoid arthritis, and it can be considered in autoimmune hepatitis after full characterization of its autoantigens.180 Synthetic peptides that compete with the autoantigen for presentation by class II MHC molecules can be used to block the first co-stimulatory signal in immunocyte activation. Soluble cytotoxic T lymphocyte antigen-4 (CTLA-4) interferes with the second co-stimulatory signal and dampens immunocyte activation.181 It has already been used to blunt immune reactivity in mismatched blood marrow recipients, and it is a powerful tool by which to manipulate the immune response.182Oral tolerance regimens induce systemic nonresponsiveness to an autoantigen by oral feedings, and this intervention has already been used in the treatment of multiple sclerosis, rheumatoid arthritis, insulin dependent diabetes mellitus, myasthenia gravis and thyroiditis.183 Low dose regimens stimulate cytokine production and suppression of the immune response, whereas high dose regimens cause clonal deletion of immunocytes and anergy. The ingested antigen is delivered directly to the liver by the portal circulation, and the treatment may be especially effective in autoimmune hepatitis once the critical epitope has been defined.184T cell vaccination can deplete clones of activated cytotoxic T lymphocytes, and it is the only site-specific intervention that has been assessed in experimental murine autoimmune hepatitis.185 Identification and precise targeting of the critical T cell clones remain challenges for this modality. Cytokine manipulations are feasible by administering drugs that disrupt intracellular signaling pathways involved in cytokine transcription or by using recombinant cytokines (such as IL-10) or monoclonal antibodies (such as anti-TNF) to alter the type of cytokine response. Similar interventions are already being evaluated in inflammatory bowel disease186 and chronic hepatitis C.187Gene therapy also has promise in the treatment of a polygenic disorder such as autoimmune hepatitis if genes can be delivered that counterbalance the over-production of certain regulatory cytokines, limit fibrosis, or promote regeneration.188
Variant syndromesCodification of the clinical criteria for the diagnosis of autoimmune hepatitis has facilitated recognition of variant syndromes.189-192 These syndromes include patients with autoimmune hepatitis and another type of chronic liver disease (overlap syndrome) or findings suggestive but non-diagnostic of autoimmune hepatitis (outlier syndrome). Overlap syndromes include patients with mixed features of autoimmune hepatitis and PBC or PSC, and outlier syndromes include patients with autoimmune cholangitis (or AMA-negative PBC) and cryptogenic chronic hepatitis. These variant conditions currently lack an established identity, official designation, and treatment strategy. Their occurrences, however, must be recognized, and they should not be assimilated into diagnoses that hide their individuality or imperil the homogeneity of the classical diseases.
The variant syndromes are important to recognize not only because they are common (occurring in 18% of patients with autoimmune liver disease), but they respond variably to corticosteroid treatment.193,194 The principal determinant of corticosteroid response is the degree of cholestasis at presentation, and this is most easily assessed by determining the magnitude of the serum alkaline phosphatase level above the upper limit of normal. Patients with variant syndromes and serum alkaline phosphatase levels greater than twofold normal are unlikely to respond to this treatment.193-196
Management of the variant syndromes is empiric and based on the predominant manifestations of the disease. Patients with autoimmune hepatitis and features of PBC who have serum alkaline phosphatase levels less than twofold normal can be treated with corticosteroids.193-195 Patients with higher serum alkaline phosphatase levels and those with florid duct lesions on histological examination are candidates for treatment with corticosteroids and ursodeoxycholic acid.196 Patients with autoimmune hepatitis and PSC lack an effective treatment, but they are candidates for a trial of therapy with prednisone and high dose ursodeoxycholic acid (15-20 mg/kg daily).193 Patients with autoimmune cholangitis can be treated with prednisone, ursodeoxycholic acid, or both depending on the serum alkaline phosphatase level.193,197 Multicenter, collaborative studies are needed to codify diagnostic criteria and establish treatment algorithms.11
SummaryAutoimmune hepatitis should be considered in all patients with acute or chronic hepatitis of undetermined cause, including patients with fulminant presentations and those who have been transplanted. Recurrent autoimmune hepatitis is possible after liver transplantation, and the disease can develop de novo in children and adults transplanted for non-autoimmune diseases. The histological spectrum of autoimmune hepatitis includes centrilobular or perivenular (Rappaport zone 3) necrosis, and background histological features of bile duct injury do not dissuade the diagnosis or alter therapy. Prednisone in combination with azathioprine is the preferred treatment in adults, and it may reduce fibrosis by suppressing inflammatory activity. New autoantibodies have promise as diagnostic and prognostic tools, and several are now available as commercial kits. Genetic factors influence disease expression and behavior, and they may be clues to region-specific etiologic agents. New treatments are evolving that promise better blanket immunosuppression and site-specific intervention. Variant syndromes are common, and they should be sought in all patients, especially in those who are refractory to corticosteroid therapy.



