



Background. The regenerative capacity of the liver is critical for proper responses to injury. Fibrin extracellular matrix (ECM) deposition is a common response to insult and contributes to inflammatory liver injury. However, the role of this matrix in hepatic regeneration has not been determined. Objective. The purpose of this study was first to determine the role of fibrin ECM in hepatic regeneration followed by the role of the fibrin-binding αvβ3 integrin in mediating this effect.
Material and methods. C57Bl/6J (WT) or PAI-1 knockout (KO) mice underwent 70% partial hepatectomy (PHx); plasma and histologic indices of regeneration were determined, as well as expression of key genes involved in hepatic regeneration.
Results. PHx promoted transient fibrin deposition by activating coagulation and concomitantly decreasing fibrinolysis. Inhibiting fibrin deposition, either by blocking thrombin (hirudin) in WT mice or by knocking out PAI-1, was associated with a decrease in hepatocyte proliferation after PHx. This strongly suggested a role for fibrin ECM in liver regeneration. To investigate if avß3 integrin mediates this action, we tested the effects of the anti-αvβ3 cyclic peptide RGDfV in animals after PHx. As was observed with inhibition of fibrin deposition, competitive inhibition of αvβ3 integrin delayed regeneration after PHx, while not affecting fibrin deposition. These effects of RGDfV correlated with impaired angiogenesis and STAT3 signaling, as well as transient endothelial dysfunction. In conclusion, these data suggest that αvβ3 integrin plays an important role in coordinating hepatocyte division during liver regeneration after PHx via crosstalk with fibrin ECM.
The liver has tremendous regenerative capacity that distinguishes it from other vital organs (e.g., the brain, heart and lungs) that are far less able to replace functional tissue. As the main detoxifying organ in the body, the liver is prone to toxic injury. Due to its regenerative properties, however, the liver is able to restore to full size and ensure survival. The liver fully regenerates within 10 d after a 70% partial hepatectomy (PHx).1 In addition to hepatocyte proliferation, there is a tightly coordinated response to complement the regenerative process, so that the entire organ can be reconstituted. Perturbations of this complex and synchronized response can impact tissue recovery from injury or damage. Indeed, it is now clear that impaired regeneration and/or restitution is critical to the chronicity of numerous hepatic diseases.
Altered homeostasis of the extracellular matrix (ECM) is well-known in liver diseases. Although the deposition of collagen ECM during fibrosis is the most familiar phenomenon, the response of the hepatic ECM to acute and chronic stress is dynamic and involves quantitative and qualitative changes in several ECM proteins. Previously work by this group and others has indicated that fibrin(ogen) ECM accumulates during acute and chronic hepatic injury and contributes to damage in several experimental models (e.g.,2,3). The hepatic ECM changes not only during injury, but also during hepatic regeneration and recovery from injury. In general, the ECM functions not only as the physical support for hepatic cells, but also modulates cell differentiation, migration, growth and apoptosis.4 However, the specific role of fibrin(ogen) ECM in hepatic regeneration is not known. A goal of this study was to therefore elucidate the specific role this protein in hepatic regeneration after PHx.
ECM fibers interact with cells through heterodimer integrin receptors, composed of α and β chains. Integrin binding transfers information from the ECM to the cell, allowing rapid and flexible responses to changes in the environment. It has been shown that integrins play a role not only in proliferation and angiogenesis, but also in inflammation and apoptosis.5–7 The integrin αvβ3 is strongly expressed on endothelial cells, hepatic stellate cells and on inflammatory cells (e.g., monocytes and macrophages8,9). Fibrin(ogen) is one of the major ligands for integrin αvβ3 and fibrin(ogen) deposition has been correlated with the expression of αvβ3.10–12 Integrin αvβ3 has been shown to play an important role in endothelial cell migration, proliferation and blood vessel formation.13,14 Integrin αvβ3 has been extensively studied in hepatic tumor growth, where it is known to play a critical role in angiogenesis and metastasis; it is therefore recognized as a key potential target in the treatment of liver cancer. However, the role of integrin αvβ3 in non-neoplastic liver growth has not been elucidated yet. Thus the second goal of this work was to determine the role of fibrin-mediated integrin signaling in hepatic regeneration.
Material and MethodsAnimals and treatmentsSix week old male C57BL/6J and PAI-1 knockout (B6.129S2-Serpine1tm1Mlg/J: PAI-1-/-) mice were purchased from The Jackson Laboratory (Bar Harbor, ME); these mice have been backcrossed at least 10 times onto C57BL/ 6, avoiding concerns regarding genetic differences between wild-type strain and the knockouts at nonspecific loci. Mice were housed in a pathogen-free barrier facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and procedures were approved by the University of Louisville’s Institutional Animal Care and Use Committee. Food and tap water were allowed ad libitum. Seventy percent partial hepatec-tomies were performed as described by Greene and Puder,15 with minor modifications. Hirudin (Refludan, Berlex, Montville, NJ, 1 mg/kg s.c.) or vehicle (saline) was given 30 min, 1.5, 4.5 and 24 h after surgery and every subsequent 24 h until sacrifice. Cyclo(-Arg-Gly-Asp-D-Phe-Val) (RGDfV; Enzo, Plymouth Meeting, PA) or vehicle (saline) was injected 1 day (1 mg/kg i.p.) prior to surgery and every 24 h (3 mg/kg i.p.) until sacrifice. Mice were anesthetized with ketamine/xylazine (100/15 mg/kg, i.m.) at select time points up to 96 h after PHx. Blood was collected from the vena cava just prior to sacrifice by exsanguination, and citrated plasma was stored at -80 °C for further analysis. Portions of liver tissue were snap-frozen in liquid nitrogen, frozen-fixed in OCT-Compound (Sakura Finetek, Torrance, CA), or were fixed in 10% neutral buffered formalin for subsequent sectioning and mounting on microscope slides.
Biochemical analysesPlasma thrombin-antithrombin (TAT) concentration was determined by enzyme-linked immunosorbent assay using a kit (Dade Behring Inc., Deerfield, IL).2,16 Plasma levels of hyaluronic acid (HA) were determined using a commercially available kit (Corgenix Inc., Broomfield, CO). Signal transducer and activator of transcription-3 (STAT3) DNA binding activity was determined using a commercially available kit from active motif (Carlsbad, CA).
Immunohistochemistry and immunofluorescenceFor detection of hepatic Proliferating Cell Nuclear Antigen (PCNA), integrin av and Cluster of Differentiation-31 (CD31), paraffin sections of liver were incubated with biotinylated PCNA antibody (DAKO, Carpenteria, CA), integrin av antibody (Millipore, Billerica, MA) or CD31 antibody (BD Pharmingen, San Diego, CA) and counterstained with hematoxylin (Sigma, St. Louis, MO). PCNA-positive stain was visualized with a DAB detection kit (DAKO, Carpenteria, CA). CD31 stains were visualized using a commercially available kit (Vectastain, Torrance, CA).
Cell cycle progression (per 1,000 hepatocytes) was estimated using PCNA staining patterns and cell morphology as described previously.17,18 The intensity and extent of CD31 staining in liver tissues were quantified by image analysis using Metamorph software (Molecular Devices, Sunnyvale, CA).
Immunofluorescent detection of fibrin, CD31 and phosphor-histone H3 was performed as previously described.2,19–21
ImmunoblotsLiver samples were homogenized in lysis buffer [20 mM Tris/Cl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% (w/v) Triton X-100], containing protease and phosphatase inhibitor cocktails (Sigma, St. Louis, MO). Samples were loaded onto SDS-polyacrylamide gels of 10% (w/v) acrylamide followed by electrophoresis and Western blotting onto polyvinylidene difluoride membranes (Hybond P, GE Healthcare, Piscataway, NJ). Primary antibodies against GAPDH and phosphorylated and total STAT3, and STAT1 (Cell Signaling Technology; Beverly, MA) were used. Bands were visualized using an Enhanced Chemiluminescence kit (Pierce, Rockford, IL) and Hyperfilm (GE Healthcare, Piscataway, NJ). Densito-metric analysis was performed using UN-SCAN-IT gel (Silk Scientific Inc., Orem, UT) software.
RNA Isolation and Real-Time rtPCRRNA extraction and real-time rtPCR was performed as described previously.2,22 Primers were purchased from Applied Biosystems (Foster City, CA); the sequences were designed to cross introns to ensure that only cDNA and not genomic DNA was amplified. The comparative CT method determines the amount of target, normalized to an endogenous reference (β-actin) and relative to a calibrator (2-ΔΔCt).
Statistical analysesResults are reported as means ± SEM (n = 4-7). Analysis of variance (ANOVA) with Bonferroni’s post-hoc test (for parametric data) or Mann-Whitney Rank Sum test (for nonparametric data) was used for the determination of statistical significance among treatment groups, as appropriate. A p value less than 0.05 was selected before the study as the level of significance.
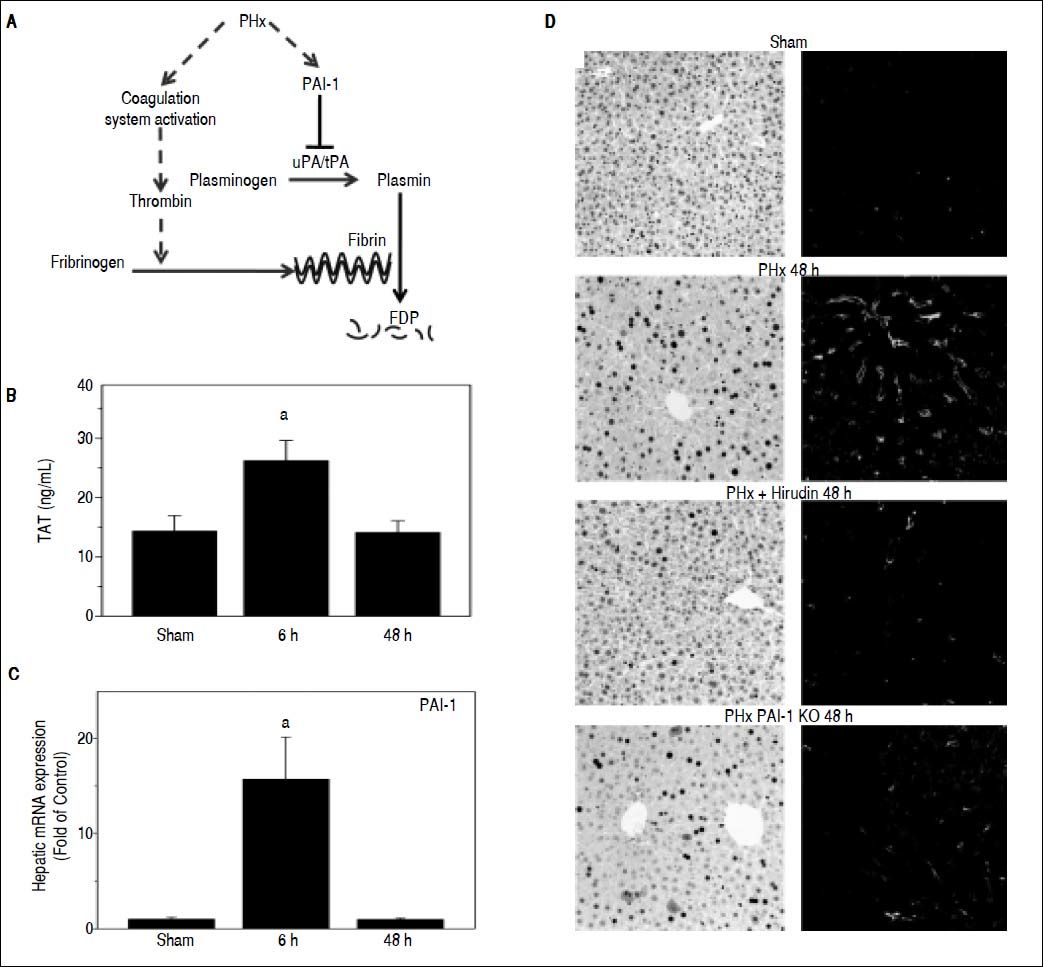
ResultsFibrin ECM homeostasis is regulated predominantly by the coagulation cascade and fibrinolysis (see Figure 1A for scheme); these pathways are not mutually exclusive and may work in tandem to mediate fibrin accumulation. To determine the effect of PHx on fibrin ECM homeostasis, indices for both coagulation (TAT dimers) and fibrinolysis (PAI-1 expression) were measured (Figures 1B and 1C). PHx significantly increased plasma TAT levels, which peaked 6 h after surgery. Analogous to TAT levels, PHx significantly increased PAI-1 maximally at 6 h after surgery. These results suggest that PHx activates coagulation, while concomitantly inhibiting fibrinolysis. To investigate the impact of these changes on fibrin levels in the hepatic sinusoids, this ECM was detected immunofluorescently. PHx caused fibrin to accumulate in sinusoidal spaces of the liver lobule as early as 6 h (data not shown) after surgery and were still elevated after 48 h (Figure 1D). To determine the role of thrombin activation in the accumulation of fibrin ECM caused by PHx, the effect of hirudin, a specific thrombin inhibitor was determined. Administration of this compound almost completely blunted the increase in fibrin ECM caused by PHx (Fig 1D). Likewise, preventing the inhibition of fibrinolysis (PAI-1-/- mice), also blunted the increase in hepatic fibrin after PHx.
. B: Plasma contents of thrombin antithrombin (TAT) were determined enzymatically and C: real-Time rtPCR was performed as described in Materials and methods. Mice were treated as described in Materials and methods. Data are means ± SEM (n = 4-6) and are expressed as fold of sham.a p < 0.05 compared to sham. D: Representative photomicrographs depicting immunohistochemical detection of hepatic PCNA and fibrin immunofuorescence 48 h after PHx are shown.")
Role of fibrin metabolism in hepatic regeneration after PHx. A: PHx activates the coagulation system including thrombin, which leads to fibrin ECM accumulation. PAI-1 inhibits the activity of the plasminogen activators uPA and tPA, blocking the activation of plasmin, thereby blunting fibrinolysis of fibrin matrices to fibrin degradation products (FDP). B: Plasma contents of thrombin antithrombin (TAT) were determined enzymatically and C: real-Time rtPCR was performed as described in Materials and methods. Mice were treated as described in Materials and methods. Data are means ± SEM (n = 4-6) and are expressed as fold of sham.a p < 0.05 compared to sham. D: Representative photomicrographs depicting immunohistochemical detection of hepatic PCNA and fibrin immunofuorescence 48 h after PHx are shown.
As mentioned in the introduction, liver can respond robustly to hepatic injury and repopulate the liver through regeneration. PHx is often used to model this response, as it focuses solely on the recovery from injury and the regenerative response is robust. Indeed, virtually all surviving hepatocytes replicate at least once after PHx. Figure 1D also shows representative photomicrographs depicting hepatocyte proliferation (PCNA staining) after PHx. As has been observed previously,23 PHx robustly increased the number of hepatocytes that were positive for PCNA staining, with ~20 and 80% of the cells in an active phase of the cell cycle (i.e., not in G0) 24 and 48 h after surgery, respectively. When fibrin accumulation was blunted with either hirudin or by knocking out PAI-1, the increase in PCNA positive cells caused by PHx was significantly blunted. The effect of hirudin was more robust than knocking-out PAI-1, in line with the relative effects of these interventions on fibrin accumulation.
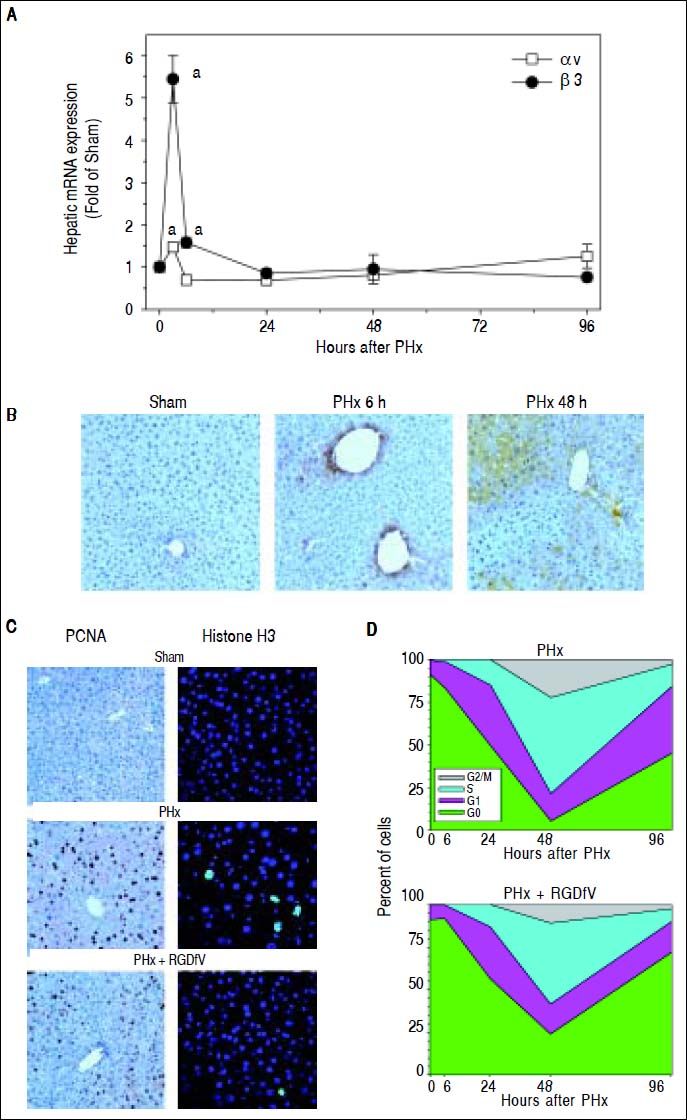
Having generated information pointing to the importance of fibrin ECM deposition in liver regeneration after PHx, the role of αvβ3 in mediating these effects was determined. To this end, the mRNA (Figure 2A) and protein (Figure 2B) expression of the subunits αv and β3 in livers of mice after PHx was first determined. Whereas PHx significantly induced hepatic integrin αv mRNA expression 3 h after surgery, β3 was also significantly induced 3 and 6 h post surgery (Figure 2A). 24 h after surgery integrin β3 mRNA levels were back to baseline (Figure 2A). Integrin av protein was still significantly increased 6, 24 and 48 h after PHx (αv IHC for 6 and 48 h timepoint, Figure 2B). This disconnect between mRNA and protein expression may reflect the lag phase between mRNA and protein levels and/or regulation of the protein levels by altered turnover.
 and are expressed as fold of sham. ap < 0.05 compared to sham. B. Representative photomicrographs depicting immunohistochemical detection of hepatic integrin αv 6 and 48 h after PHx are shown. C. Representative photomicrographs depicting immunohistochemical detection of hepatic PCNA and Histone H3 immunofluorescence at the 48 h timepoint are shown. D. Cell cycle progression (per 1,000 hepatocytes) was estimated at different time points. Cell counting data are means ± SEM (n = 4-6).")
Integrin αββ3 is critically involved in hepatocyte proliferation after PHx. Mice were treated and A: real-time rtPCRs for integrins αv and β3 were performed as described in Materials and methods. Data are means ± SEM (n = 4-6) and are expressed as fold of sham. ap < 0.05 compared to sham. B. Representative photomicrographs depicting immunohistochemical detection of hepatic integrin αv 6 and 48 h after PHx are shown. C. Representative photomicrographs depicting immunohistochemical detection of hepatic PCNA and Histone H3 immunofluorescence at the 48 h timepoint are shown. D. Cell cycle progression (per 1,000 hepatocytes) was estimated at different time points. Cell counting data are means ± SEM (n = 4-6).
To determine if integrin αvβ3 contributes to hepatic regeneration, the impact of blocking the interaction of this integrin with ECM proteins [e.g., fibrin(ogen)] was determined with RGDfV administration. Figure 2C shows representative photomicrographs of the 48 h timepoint depicting hepatocyte proliferation (PCNA staining) and Figure 2D shows graphical comparison of PCNA staining between PHx and PHx + RGDfV at all timepoints. As described above (Figure 1), PHx robustly increased the number of dividing hepatocytes as early as 6 h after surgery with a peak at 48 h (Figure 2D). RGDfV significantly decreased the number of PCNA-positive cells (i.e., not G0) after PHx at all timepoints. A delayed entry and progression through the cell cycle was shown for the RGDfV group (Figure 2D). Furthermore, figure 2 shows representative photomicrographs of the 48 h timepoint stained for phospho-histone H3 immunofluorescence, as an index of mitosis. PHx robustly increased the number of mitotic hepatocytes (Figures 2C and 2D). This increase caused by PHx was significantly blunted by RGDfV administration (Figures 2C and 2D).
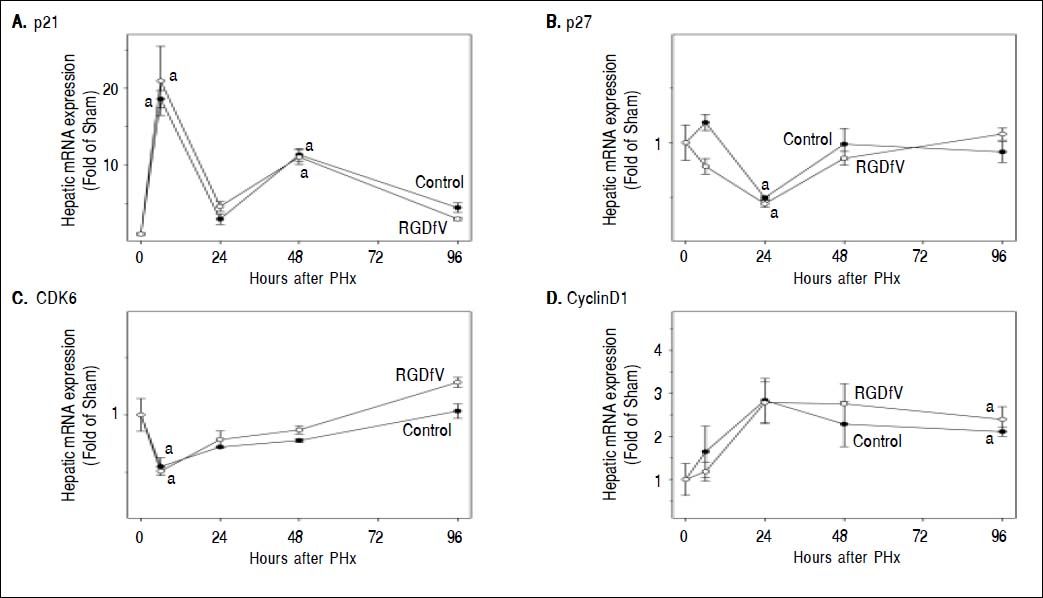
Inhibition of cell proliferation after PHx by RGDfV led to the investigation of potential mechanisms by which this occurred. Primary attention was focused on key mechanisms related to cell cycle control, angiogenesis and cytokine signaling. Therefore, hepatic mRNA levels of key inducers (e.g., CDK6 and cyclin D1) and inhibitors (e.g., p21 and p27) of the cell cycle were determined by rtPCR (Figure 3). As described previously,23 PHx significantly altered the expression of all these mediators with different temporal patterns. For example, PHx increased the expression p21 and cyclin D1 rapidly, and levels slowly returned to basal at later times after PHx. In contrast, PHx decreased the expression of p27 and CDK6 at early timepoints, and then expression tended to return to baseline by the end of the experiment. RGDfV administration did not significantly impact the expression of these genes at any timepoint tested (Figure 3).
 and are expressed as fold of sham.a, p < 0.05 compared to sham;b, p < 0.05 compared to PHx.")
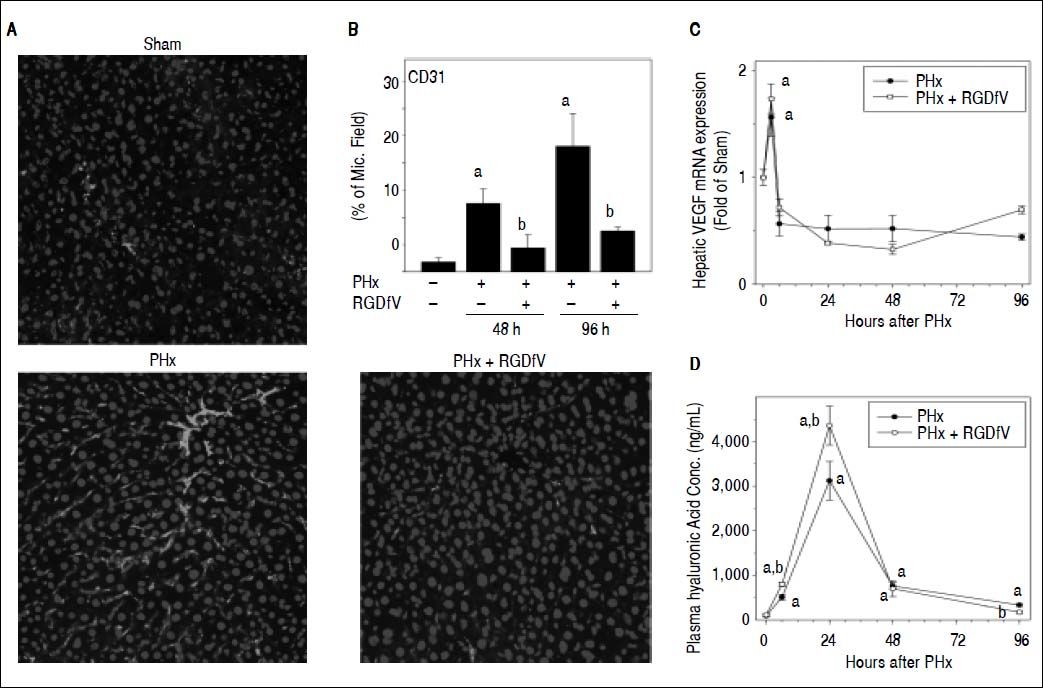
As mentioned in the Introduction, whereas hepatocyte division is necessary, it is not sufficient for complete liver regeneration. For example, liver regeneration requires neoangiogenesis to support expansion of these new cells. Liver regeneration is therefore highly regulated by endothelial cell function and expansion, especially at later timepoints. Impairment of this response could block hepatic regeneration, even under conditions where expression of cell cycle genes are unaffected (Figure 3). Given the known role of αvβ3 integrins in basal and pathologic endothelial function and angiogenesis,13,14 the effect of RGDfV administration on indices of these functions were determined. CD31 immunofluorescence as a marker of endothelial cells/angiogenesis is shown in Figure 4A. PHx increased CD31 immunofluorescence at later timepoints (i.e., > 48 h); this increase by PHx was significantly attenuated by RGDfV (Figure 4B). HA is almost exclusively metabolized by endothelial cells and plasma levels are crude indices of endothelial function. PHx robustly increased plasma levels HA, which was increased by ~30% by RGDfV administration at the peak time (24 h; Figure 4D). These effects of RGDfV endothelial cells was independent of changes in expression of the potent endothelial mitogen, vascular endothelial growth factor (VEGF), which was increased by PHx, but not affected by RGDfV (Figure 4C).
 image analysis of CD31 positive staining are shown. C. A timecourse for hepatic VEGF expression is shown. Panel D depicts plasma levels of hyaluronic acid. Data are means ± SEM (n = 4-6) and are expressed as % of microscope field for CD31 (B), as fold of sham for mRNA expression (C) and as ng/mL for plasma hyaluronic acid concentration (D). ap < 0.05 compared to sham; bp < 0.05 compared to PHx.")
Effect of integrin αvβ3 inhibition on endothelial cell function after PHx. Mice were treated and real-Time rtPCR was performed as described in Materials and methods. A. Representative photomicrographs of mouse livers depicting CD31 immunofluorescence and (B) image analysis of CD31 positive staining are shown. C. A timecourse for hepatic VEGF expression is shown. Panel D depicts plasma levels of hyaluronic acid. Data are means ± SEM (n = 4-6) and are expressed as % of microscope field for CD31 (B), as fold of sham for mRNA expression (C) and as ng/mL for plasma hyaluronic acid concentration (D). ap < 0.05 compared to sham; bp < 0.05 compared to PHx.
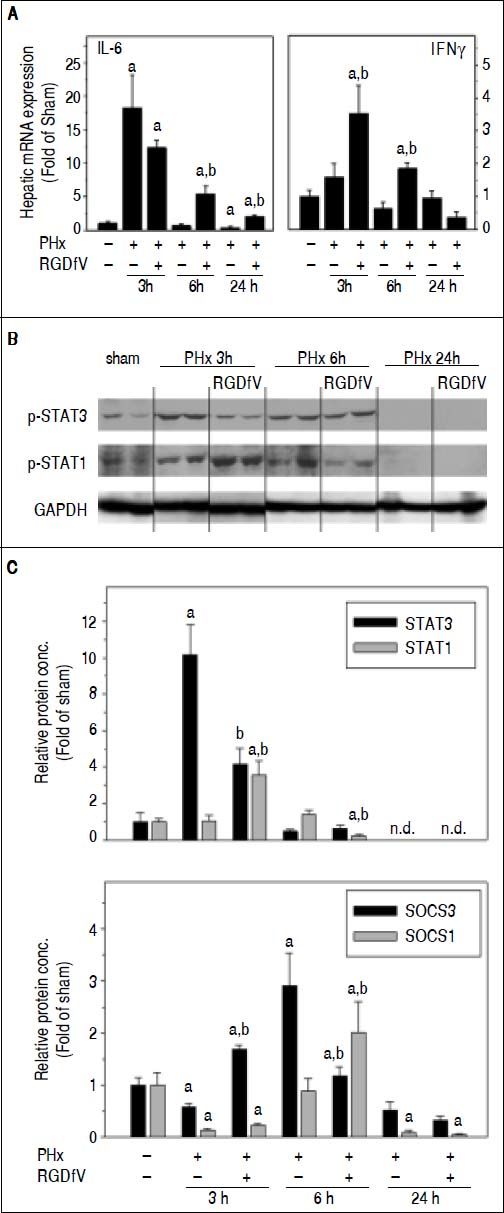
Although impaired angiogenesis (Figure 4) may explain delays in regeneration caused by RGDfV at later timepoints after PHx, the earlier effects of RGDfV (up to 24 h) on regeneration appeared independent of these variables. It is well-known that the cytokine IL-6 plays critical roles in the early initiation and progression of liver regeneration after PHx.24 Indeed, the expression of IL-6 was strongly increased as early as 3 h after PHx, but returned rapidly to baseline by 24 h (Figure 5A). RGDfV did not significantly inhibit expression of this mitogen; indeed, the mRNA expression of IL-6 was actually increased by RGDfV administration at the 6 h timepoint (Figure 5A). Similar effects were observed for IL-6 protein levels (sham, 2.8 ± 0.4 pg/mL) at the 3h (PHx, 9.8 ± 1.8; PHx ± RGDfV 13.9 ± 0.4 pg/mL, p < 0.05) and 6 h (PHx, 5.6 ± 1.0; PHx ± RGDfV, 8.6 ± 1.1 pg/mL, p < 0.05). IL-6 is a major inducer of the STAT3 signaling pathway, which is critical for hepatocyte survival and liver regeneration.25,26 Indeed, STAT3 phosphorylation was rapidly and robustly increased 3 h after PHx, but had returned to baseline by 6 h (Figure 5B). Even though RGDfV increased hepatic IL-6 levels after PHx (Figure 5A), the increase in STAT3 phosphorylation caused by PHx was attenuated > 2-fold by RGDfV administration (Figure 5B). RGDfV did not affect the expression of other mitogens involved in the early response to PHx (e.g., TNFa and HGF; not shown).
 and Western Blot analysis (B) were performed as described in Materials and methods. Data are means ± SEM (n = 4-6) and are expressed as fold of sham. ap < 0.05 compared to sham; bp < 0.05 compared to PHx.")
Effect of PHx and RGDfV on the expression of IL-6/STAT3 and IFNy/STAT1 pathways. Real-Time rtPCR (A, C) and Western Blot analysis (B) were performed as described in Materials and methods. Data are means ± SEM (n = 4-6) and are expressed as fold of sham. ap < 0.05 compared to sham; bp < 0.05 compared to PHx.
The mitogenic effects of IL-6 via STAT3 signaling are opposed by the cytokine IFNy, via STAT1 activation during liver regeneration27,28 (Figure 6). The increase in expression of IFNy caused by PHx was significantly enhanced by RGDfV 3 and 6 h after PHx (Figure 5A). This increase in IFNy correlated with enhanced STAT1 phosphorylation in the PHx + RGDfV group (Figure 5B). STAT1-dependent signaling increases the expression of suppressor of cytokine signaling (SOCS), which subsequently blunt STAT3 signaling.29 Indeed, whereas expression of SOCS3 was attenuated in the PHx group 3 h after surgery, it was significantly enhanced in the PHx + RGDfV group (Figure 5C). Likewise, the expression of SOCS1 was significantly enhanced in the PHx + RGDfV group at the 6 h timepoint (Figure 5C).

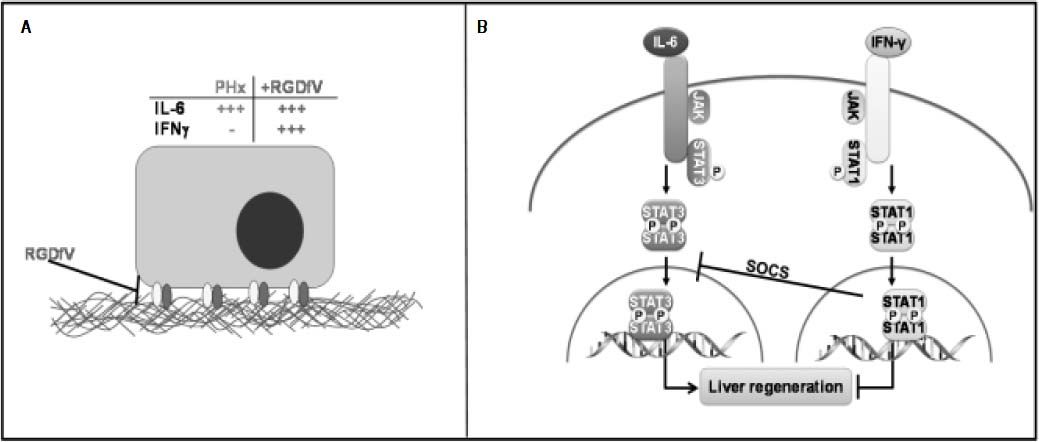
Working hypothesis. In this model, the increase in integrin αvβ3 correlates with an increase in hepatic regeneration and angiogenesis. Both are blunted by inhibiting integrin αvβ3 with RGDfV. IL-6 induction has been shown to be crucial for liver regeneration due to PHx. In this model, however IL-6 induction does not correlate with an increase in proliferation and angiogenesis in the RGDfV group. Studies have shown that, whereas IL-6-induced activation of hepatic STAT3 after PHx and is critica in the promotion of liver regeneration, IFN-γ-induced STAT1 activation blocks liver regeneration after PHx.27,43 Indeed, in this model IFN-γ induction as well as STAT1 activation concomitant with a decrease in STAT3 activation.
Regeneration of the liver is unique among the solid organs of the body and has been well recognized since ancient times.1 Nevertheless the mechanisms behind this effect remain incompletely understood. These gaps in our knowledge impact procedures like liver transplantation, liver resection or partial hepatectomy where regeneration plays a key role. Furthermore, better understanding of the mechanisms of liver regeneration can shed light on pathogenic processes that involve proliferation and/or remodeling, such as hepatic fibrosis and carcinogenesis.
The purpose of the current study was to investigate the potential interaction between fibrin ECM and hepatic regeneration. Previous work has partially characterized the changes in the hepatic ECM caused by PHx.30 That previous study demonstrated that “fibrinogen degradation products” (i.e., fibrin) accumulate in the liver after PHx. Since the level of fibrin ECM can be regulated both at the level of deposition as well as at the level of fibrinolysis (Figure 1A; see references 31 and 32 for review), the mechanism(s) by which fibrin ECM accumulates after PHx was unclear. Here, blocking the coagulation cascade (hirudin) and genetic deletion of PAI-1 both prevented the increase in fibrin ECM caused by PHx (Figure 1), indicating that both pathways likely contribute to transient accumulation of fibrin ECM in the liver.
As mentioned in the Introduction, fibrin ECM accumulation is an acute phase response in several models of hepatic injury (see reference 33 for review). Fibrin clots disrupt the flow of blood within the hepatic parenchyma (i.e., hemostasis) and enhance inflammation.33 Indeed, under most conditions, blocking fibrin ECM accumulation is protective against both acute and chronic liver injury.33 Two notable exceptions are that PAI-1 deficient mice are more sensitive to liver injury caused by acetaminophen34 and carbon tetrachloride.17 In both studies, PAI-1 deletion and subsequent inhibition of fibrin ECM accumulation correlated with an impaired regenerative response.17,34 These studies suggest a novel function for PAI-1 via fibrin ECM, in hepatic regeneration. However, it is difficult to determine cause and effect relationships for models in which regeneration is driven by toxic insult to the organ. The data shown here in the PHx model therefore more clearly demonstrate a role of transient fibrin ECM accumulation in liver regeneration (Figure 1).
Cells interact with fibrin(ogen) via multiple surface receptors. A major fibrin-binding receptor in hepatic cells is integrin αvβ3, which promotes fibrin binding through its RGD domain. Integrin αvβ3 has an established role in pathogenic neoangiogenesis in the liver (e.g., hepatocellular carcinoma).5–7 However, the role of this integrin in nonneoplastic angiogenesis (i.e., after PHx) was unclear. It was shown here that PHx increased integrin αvβ3 expression (Figure 2) and that blocking integrin αvβ3 with a specific antagonist (RGDfV) impaired hepatic regeneration after PHx in mice (Figure 2C). The cell cycle is regulated at the level of initiation, but also at the level of progression, with the G1/S and the G2/M transitions being key checkpoints.35 Here, RGDfV significantly decreased the number of PCNA-positive cells after PHx at all timepoints, which is indicative of a delayed entry into and progression through the cell cycle (Figure 2D). These transitions are regulated, in part, by cell cycle mediators (e.g., CDK6 and cyclin D1) and inhibitors (e.g., p21 and p27). However, the effect of RGDfV impairing hepatic regeneration did not correlate with altered expression of cell cycle control genes (Figure 3). These results suggest that other mechanisms contribute to the inhibitory effect of RGDfV observed here (see below).
As discussed in the Introduction, successful liver regeneration relies not only on parenchymal cell division, but also on processes that support the formation of the hepatic unit. It is known that liver regeneration is also dependent on angiogenesis. For example, studies have shown that antiangiogenic agents, e.g. angiostatin decrease liver regeneration after partial hepatectomy in mice.36 Here, RGDfV significantly blunted the proangiogenic response caused by PHx, determined by immunofluorescent staining of endothelial cell marker CD31 (Figure 4). Integrin αvβ3 has been shown to be highly expressed in activated endothelial cells, favoring angiogenesis.37–40 In the current study, RGD-fV significantly exacerbated the PHx-induced increase in plasma levels of HA, indicating enhanced transient endothelial dysfunction by RGDfV under these conditions (Figure 4D). These effects appeared ’downstream’ of fibrin accumulation, as RGDfV administration did not affect the accumulation of fibrin caused by PHx (not shown).
The regenerative response after PHx is mediated by numerous signals, with IL-6 being a critical initiating mitogen.24 IL-6 regulates immediate-early genes activated in the liver after PHx, and disruption of IL-6 or STAT3 impairs liver regeneration.25,41,42 Importantly, some of the mechanisms by which IL-6 mediates liver regeneration appear to be independent of regulation of cell cycle control genes,25 analogous to results observed here with RGDfV (Figure 3). In the current study, however, IL-6 induction by PHx was actually enhanced by RGDfV (Figure 5), yet this effect correlated with impaired regeneration in this group (Figure 2C). Indeed STAT3 phosphorylation was blunted by RGDfV after PHx and the expression of the STAT3-dependent gene VEGF was not enhanced (Figure 4C). This impairment of STAT3 phosphorylation (Figure 5B), despite increased IL-6 levels, correlated with an increase in STAT1 phosphorylation (Figure 5B). STAT1 and STAT3 play opposing roles in many aspects of liver injury and repair. Studies have shown that, whereas IL-6-induced activation of hepatic STAT3 after PHx and is critical in the promotion of liver regeneration, IFNγ-induced STAT1 activation blocks liver regeneration after PHx.27,43 Indeed, here IFNγ induction caused by PHx was enhanced by RGDfV (Figure 5A). Furthermore, the PHx-induced increase in mRNA expression of SOCS3 and SOCS1, STAT1-dependent genes and STAT3 inhibitors are blunted in the RGDfV group (Figure 5C). These data suggest that STAT3 is not fully activated and/or does not translocate to the nucleus under these conditions. Indeed, there was no detectable STAT3 DNA binding activity in nuclear extracts of liver tissue (data not shown).
ConclusionsTaken together, these results suggest that proliferation is regulated, at least in part, by interaction of cellular integrins with RGD-containing ECM (e.g., fibrin). The effects observed here may be explained by the well-known role of integrins in mediating anchorage-dependent growth in non-neoplastic cells.44 These results suggest that disrupting this interaction shifts from pro-proliferative signaling (via IL-6) to anti-proliferative/pro-apoptotic signaling (via IFNγ) at early timepoints after PHx. Furthermore, integrin αvβ3 mediated angiogenesis likely is critical to hepatic regeneration at the later timepoints (e.g., 48 and 96 h), analogous to its known effect in hepatic neoplasms. The results of this study also support previous findings in toxic liver injury models,17,34 in that blocking fibrin ECM accumulation can blunt hepatic regeneration in the liver. The results here suggest that inhibiting fibrin ECM accumulation via thrombin inhibition (Hirudin) or in PAI-1 knockout mice is detrimental to hepatic regeneration after PHx. These findings are somewhat surprising given the assumed antiproliferative effect of PAI-1 mediated via inhibiting maturation of hepatocyte growth factor (HGF). However, these may yield insight into the “PAI-1 paradox” in cancer, in which high levels of PAI-1 correlate with more aggressive, more rapidly dividing neoplasms.45
Abbreviations- •
ANOVA: analysis of variance.
- •
AUC: area under the curve.
- •
CD: cluster of differentiation.
- •
CDK: cyclin-dependent kinase.
- •
DAPI: 4’,6-diamidino-2-phenylindole, dihydrochloride.
- •
ECM: extracellular matrix.
- •
HA: hyaluronic acid.
- •
IFN: interferon.
- •
IL: interleukin.
- •
PAI-1: plasminogen activator inhibitor-1.
- •
PCNA: proliferating cell nuclear antigen.
- •
PHx: partial hepatectomy.
- •
RGDfV: cyclo(-Arg-Gly-Asp-D-Phe-Val).
- •
rtPCR: reverse transcription polymerase chain reaction.
- •
SOCS: suppressor of cytokine signaling.
- •
STAT: signal transducer and activator of transcription.
- •
TAT: thrombin-antithrombin.
- •
VEGF: vascular endothelial growth factor.
Supported, in part, by grants from NIAAA (R01 AA003624 and R01 AA021978).
Concept and design, analysis and/or interpretation of data (JI Beier, L Guo, S Joshi-Barve, JD Ritzenthaler, J Roman and GE Arteel); critical writing or revising the intellectual content (JI Beier, J Roman and GE Arteel); final approval of the version to be published (JI Beier, L Guo, S Joshi-Barve, JD Ritzenthaler, J Roman and GE Arteel).



