





Chronic hepatitis B (CHB) is a global epidemic disease that results from hepatitis B virus (HBV) infection and may progress to liver cirrhosis. The relationship between hepatitis B virus-related cirrhosis (HBV-RC) and gut microbiota dysbiosis is still unclear. The aim of this study is to elucidate the compositional and functional characteristics of the gut microbiota in the patients with liver cirrhosis and healthy individuals.
Materials and methodsWe analyzed the gut microbiome in patients with HBV-RC and healthy individuals by 16S rRNA sequencing and metagenomic sequencing of fecal samples. A total of 113 genera, 85 families, 57 orders, 44 classes and 21 phyla were performed.
ResultsOur results suggests that the composition of the gut microbiota had changed in the early stages of cirrhosis. We further identified more than 17 genera with different richness in compensated and decompensated cirrhosis groups. PICRUSt analysis showed that changes in bacterial composition can lead to significant changes in gene function, which may be one of the causes of liver cirrhosis.
ConclusionOur study demonstrated that the composition of gut microbiota changed at different phases of HBV-RC. Gut microbiome transformation may be a biological factor in the progression of cirrhosis.
The situation of hepatitis B virus infection is still very serious. To date, more than 250 million people worldwide are chronically infected with the Hepatitis B virus (HBV) [1]. Liver cirrhosis is the pathologic end stage of chronic liver disease. Patients with chronic hepatitis B (CHB) are at a high risk of developing cirrhosis. Therefore, dynamic monitoring of disease progression in patients with hepatitis B virus-related cirrhosis (HBV-RC) is necessary.
There are three factors in the aetiology of HBV-RC: genetic (such as single nucleotide polymorphism), virus (serotypes, genotypes, gene subtypes, quasispecies, etc) and environmental factors [2]. The gut microbiota, containing 100–150 times more genes than the human genome and carrying more than 100 trillion microorganisms, plays a vital role in many aspects of the pathophysiology of intestinal and extra intestinal diseases, including immunity, neurophysiology and metabolism as well as liver fibrosis and cirrhosis [3].
As known to all, the liver and gut are closely related. The gut depends on the bile secreted by the liver for normal digestion and absorption. The liver gets about 70% of its blood supply from the gut. There is also a strong link between the liver and gut microbiota. As a detoxification organ, the liver removes toxins, harmful enteric bacteria and fungi from the gut [3,4]. The normal intestinal microflora assists in the absorption, decomposition and metabolism of various nutrients, which is an important factor to guarantee the normal operation of the liver. When liver function is impaired, such as hepatitis and cirrhosis, the gut microbiome changes dramatically. The intestinal mucosal immune system composed of intestinal microflora and intestinal immune cells will be damaged, resulting in dysbacteriosis [5,6].
An increasing body of evidence suggested that gut microbiota had played an important role in the pathogenesis of liver diseases in recent years [3,4]. Some study had indicated the effect of HBV infection on gut microbiota and the relationship between cirrhosis progression and alteration of gut microbiota [7]. However, there are few studies on the interaction between gut microbiota dysbiosis and HBV-infection-induced cirrhosis patients. In this study, high-throughput 16s rDNA gene sequencing was performed to investigate the profile of gut microbiota between patients with decompensated and compensated cirrhosis.
2Materials and methods2.1Participant informationThe study was conducted in The First Affiliated Hospital of Fujian Medical University between 1 February 2019 and 30 December 2020 (Fig. S1). This study was approved by the Ethics Approval Committee of research and Clinical Technology Application Branch of the Ethics Committee of the First Affiliated Hospital of Fujian Medical University and informed consent was obtained prior to enrollment.
The HBV-RC patients were from the Center of Liver Diseases of the First Affiliated Hospital of Fujian Medical University. The healthy person includes the healthy students studied at Fujian Medical University and healthy volunteers who visited the physical examination center of the First Affiliated Hospital of Fujian Medical University. The HBV-RC patients were diagnosed to be positive for HBsAg for at least 6 months; the diagnosis of decompensated cirrhosis and compensated cirrhosis was made according to the ‘Guidelines for diagnosis and Treatment of cirrhosis (2020 Version)’. Groups was determined using the Child-Turcotte-Pugh (CTP) scoring system, in which class A was defined as compensated cirrhosis (CC) group and classes B and C were defined as decompensated cirrhosis (DC) group.
The exclusion criteria included: (1) Patients not treated with antibiotics; (2) co-infection with hepatitis C virus, hepatitis D virus, human immunodeficiency virus or decompensated liver disease, pregnancy and alcoholism within 1 year before the treatment; (3) history of therapy with systemic corticosteroids, antineoplastic or immunomodulator drugs.
2.2Laboratory measurementsBlood biochemical values including alanine aminotransferase (ALT), aspartate aminotransferase (AST), total protein (TP), triglycerides (TG), high-density lipoprotein (HDL), low-density lipoprotein (LDL) and alkaline phosphatase (ALP) was determined by automated biochemical technique (Siemens Healthcare Diagnostics). Platelet count (PC) was tested on Siemens ADVIA 2120 Hematology Analyzer and prothrombin time (PT) was measured by the Sysmex CS-5100 Hemostasis System,. HBV serum markers including HBsAg, anti-HBs, HBeAg, anti-HBe and anti-HBc were performed on the automatic immune fluorescence analyzer Abbott Type i4000 (Abbott Laboratories). Serum levels of HBV DNA was quantified by qPCR (QuantStudio™ Dx Real-Time PCR System).
2.3Collection of fecal samples and extraction of DNATotal bacterial DNA was isolated using a QIAamp Fast DNA stool mini kit (Qiagen) according to the manufacturer's instruction. The fresh fecal samples were collected in sterile plastic tubes and stored at -80°C immediately.
2.416S rDNA gene sequencingTo analyze the gut microbiota, fecal samples of the participants were collected for 16s rDNA sequencing. The forward primer: (5’-CTTTCCCTACACGAC-3’) and reverse primer: (5’-TGGAGTTCAGACGTGT-3’) and sample-specific barcode sequence were used to amplify the V3-V4 highly variable region of the 16S rDNA gene sequence. The 16s rDNA was PCR-amplified by Illumina HiSeq platform (Illumina).
2.5Bioinformatic and statistical analysis16s rDNA gene sequencing data were analyzed by the Quantitative Insights into Microbial Ecology platform (QIIME). Bacterial diversity was determined by sampling-based OTUs analysis and presented by Shannon index, Simpson index Chao1 and ACE, which was calculated using R program package. Principal coordinates analysis (PCoA) was conducted by R package (http://www.r-project.org/) to display microbiome space between samples. The obtained splicing sequences were called raw tags. Operational taxonomic units (OTUs) were picked using closed reference OTU picking based on ≥ 97% similarity threshold using UCLUST algorithm after removal of singletons.
Bacterial taxonomic analyses and comparison including bacterial phylum and genus were conducted between two groups using Wilcoxon rank sum test. Faecal microbial characterization were analyzed by linear discriminant analysis (LDA) effect size (LEfSe) method (http://huttenhower.sph.harvard.edu/lefse/). Based on the normalized relative abundance matrix, features with significantly different abundances between assigned taxa were determined by LEfSe with Kruskal-Wallis rank sum test (p < 0.05) and LDA was used to assess the effect size of each feature.
Kruskal-Wallis rank sum test, Wilcoxon rank sum test, Chisquared, nonparametric and Student's t test were evaluate the differences among the three groups. Statistical analyses were performed using SPSS software for Mac OS X (version 23.0; SPSS Inc) and R software for Mac OS X (version 3.3.3; R Foundation for Statistical Computing, Vienna, Austria). A P value < 0.05 was considered significant.
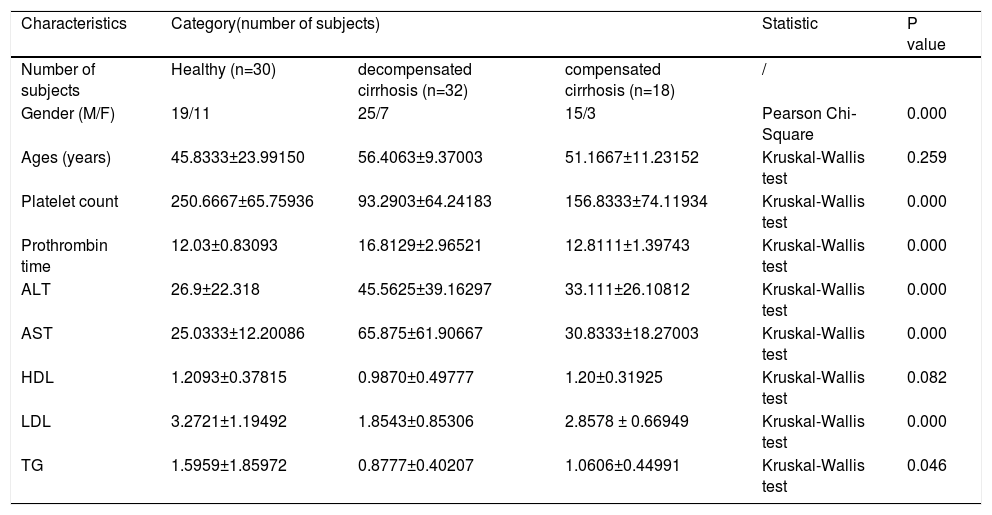
3Results3.1Clinical characteristics of the participants30 healthy individuals were recruited in this study. The HBV-RC patients were divided into two groups: decompensated cirrhosis (DC) and compensated cirrhosis (CC). The clinical characteristics of participants were shown in Table 1. Serum levels of ALT, AST were significantly increased, while the concentration of TG was markedly decreased in HBV-RC patients.
The demographics and clinical characteristics of patients and healthy subjects.
Note: Continuous variables were expressed as means ± standard deviation. Superscript letters indicated a significant difference (P < 0.05). Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; HDL, high-density lipoprotein; LDL, low-density lipoprotein; NA, not applicable; TG, triglycerides.
A total of 80 fecal samples were obtained from healthy subjects and HBV-RC patients. These sample were sequenced to generate V3-V4 16S rDNA gene profiles. A total of 1688553, 1875037, 1021045 sequences were acquired for the DC, CC and healthy groups respectively after excluding low-quality reads. The average sequence length, the maximum length and the minimum length were 416.07 bp, 453 bp and 180 bp, respectively (Fig. S2).
The depths of sequencing were measured by drawing the rarefaction curve from the Shannon index. Most samples reached plateaus, indicating that sequencing depth was adequate (Fig. S3). The species accumulation curve also showed a saturation phase and a plateau, which indicated that the size of sample was large enough to capture the entire microbiome structure (Fig. S4).
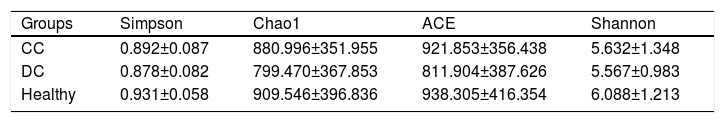
3.3The analysis of α-diversity indexesThe α-diversity indexes including Simpson, Chao1, ACE and Shannon are shown in Table 2. Although these data were higher in patients with compensated cirrhosis than in those with decompensated cirrhosis, these differences did not reach the significance level by t test.
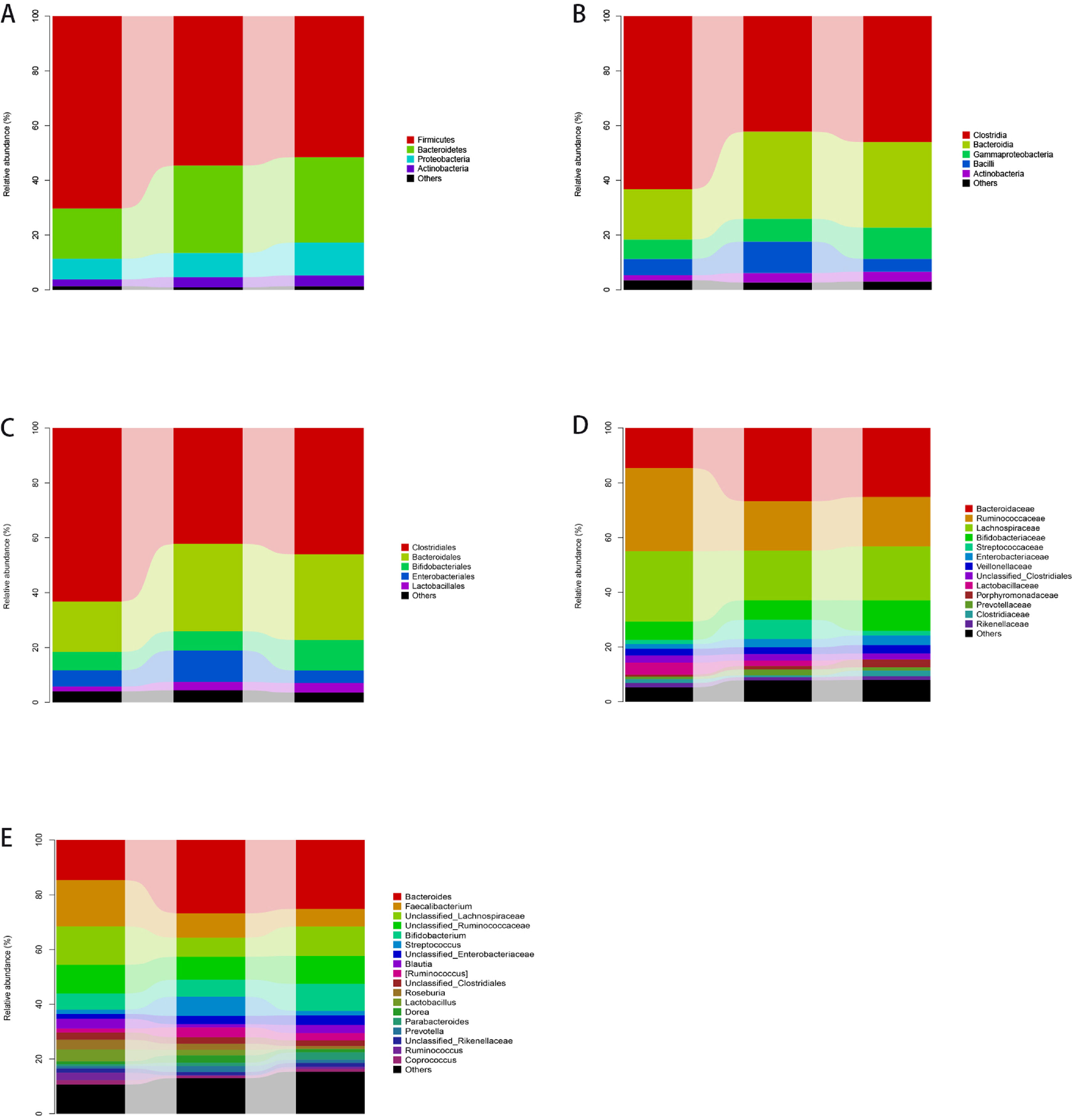
3.4The taxonomic distributions of the predominant bacteria at different levelsA total of 113 genera, 85 families, 57 orders, 44 classes, and 21 phyla were detected from stool specimens. The bacterial abundance and distribution of the dominant bacteria at different levels (relative abundance > 1% of the total sequences) is shown in Fig. 1. Some of the most abundant phyla were Firmicutes (57.0%), Bacteroidetes (28.6%), Proteobacteria (9.7%), Actinobacteria (3.5%), Fusobacteria (0.8%) and Verrucomicrobia (0.3%), together comprising 92.1% of the total sequences.

The abundance of gut microbiota at different taxonomic levels. Fig. 1A–E show the distribution of the predominant bacteria at phylum, class, order, family and genus levels respectively.
The most abundant genera were Bacteroides (23.4%), Lachnospiraceae (10%), Faecalibacterium (9.8%), Ruminococcaceae (9.5%), Enterobacteriaceae (7.5%), Streptococcus (3.8%), Bifidobacterium (2.9%), Ruminococcus (2.7%), Blautia (2.4%), Clostridiales (2.3%), Roseburia (2.1%), Lactobacillus (1.8%), Dorea (1.7%), Parabacteroides (1.7%), Prevotella (1.5%), Rikenellaceae (1.3%), Coprococcus (1.1%), Enterococcus (1.1%) and Ruminococcus (1.0%), together accounting for 87.6% of the total sequences. MEGAN software was used to map OTU abundance information and taxonomic composition data contained in each sample to the microbial clarification ranking tree (NCBI Taxonomy), and analysis was showed in Fig. S5. Fig. S6 shows a heat map indicating significantly expressed genera. It is clear that the abundance useful Bifidobacterium and Lactobacillus were reduced and the abundance of harmful Enterobacteriaceae was increased in cirrhosis patients compared to healthy controls. The abundance of harmful Steptococcus and Ruminococcus was higher in patients with decompensated cirrhosis than in those with compensated cirrhosis, suggesting that the balance of gut microbiota in these patients was severely disrupted.
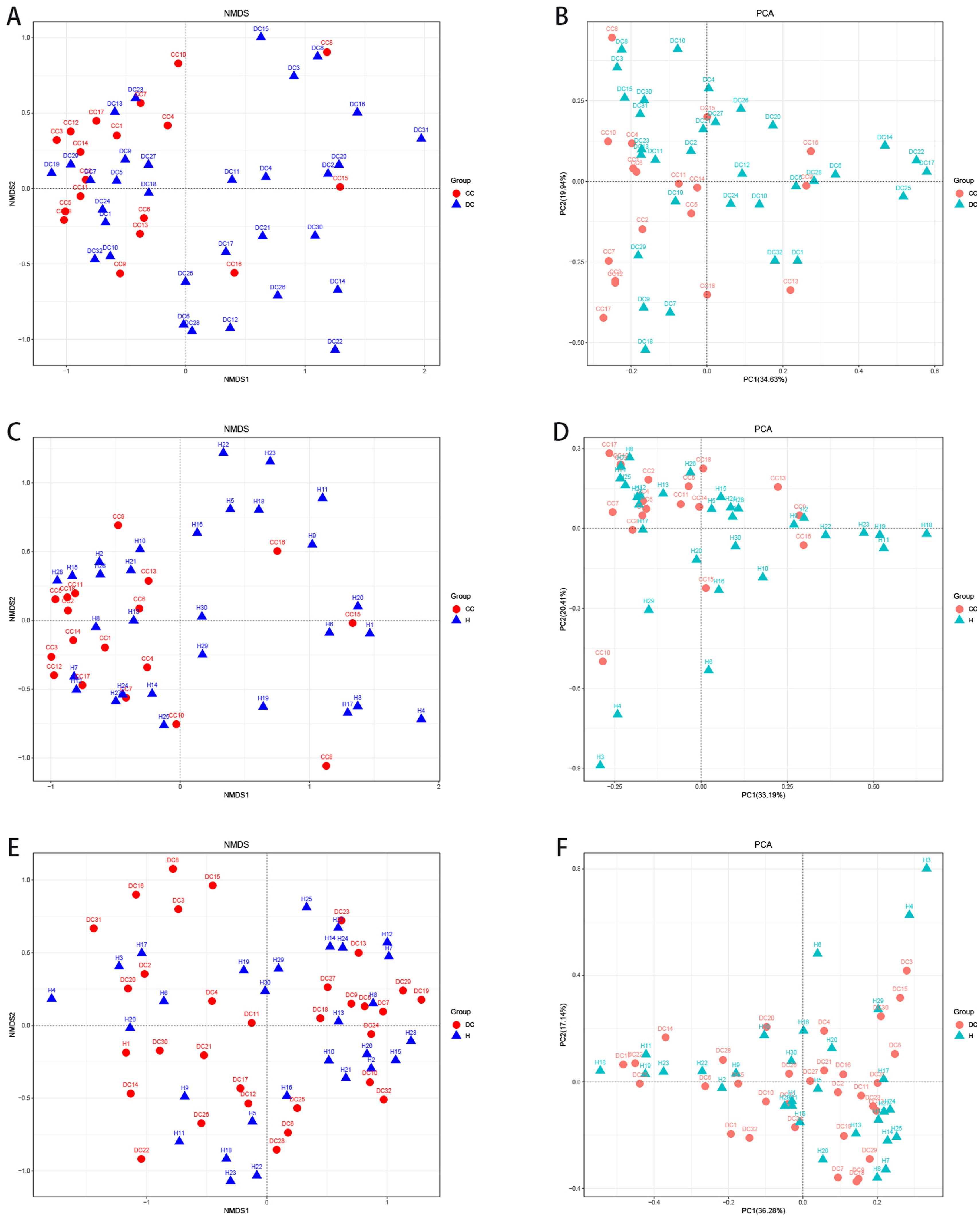
3.5The analysis of β-diversity indexesThe β-diversity analysis using the weighted UniFrac method and principal coordinate analysis showed a separate clustering of HBV-RC patients and healthy individuals covering decompensated cirrhosis and compensated cirhosis (Fig. 2). As shown in Fig. 2, the gut microbiota of HBV-RC patients varied substantially while those of healthy individuals were more consistent.
 analysis. B and D, principal component analysis on the relative abundance. Each sample was represented by a dot.")
The β-diversity analysis. A and C, comparing sample distributions belonging to different liver cirrhosis subgroups by using weighted nonmetric multidimensional scaling (NMDS) analysis. B and D, principal component analysis on the relative abundance. Each sample was represented by a dot.
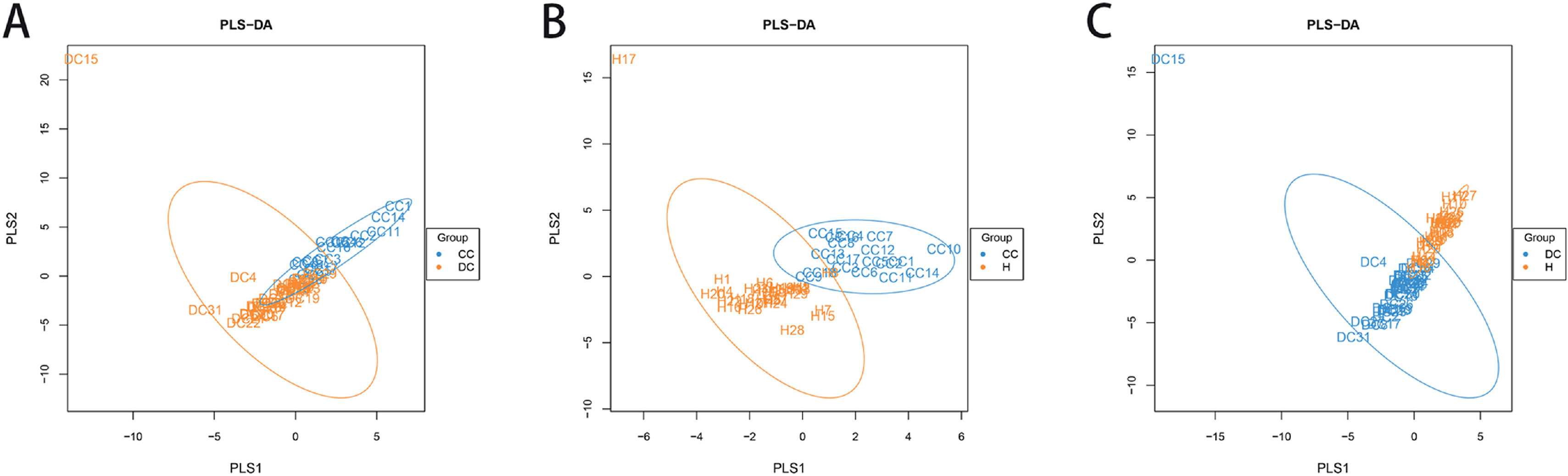
A PLS-DA, which was based on partial least squares regression model, discriminant analysis was performed on the community structure data according to the given sample distribution/grouping information., As shown in Fig. 3, there were structural differences in the gut bacterial community structure among the groups, indicating that the classification model was effective.
3.7Differential gut microbiota compositions between different stage of cirrhosis and. healthy
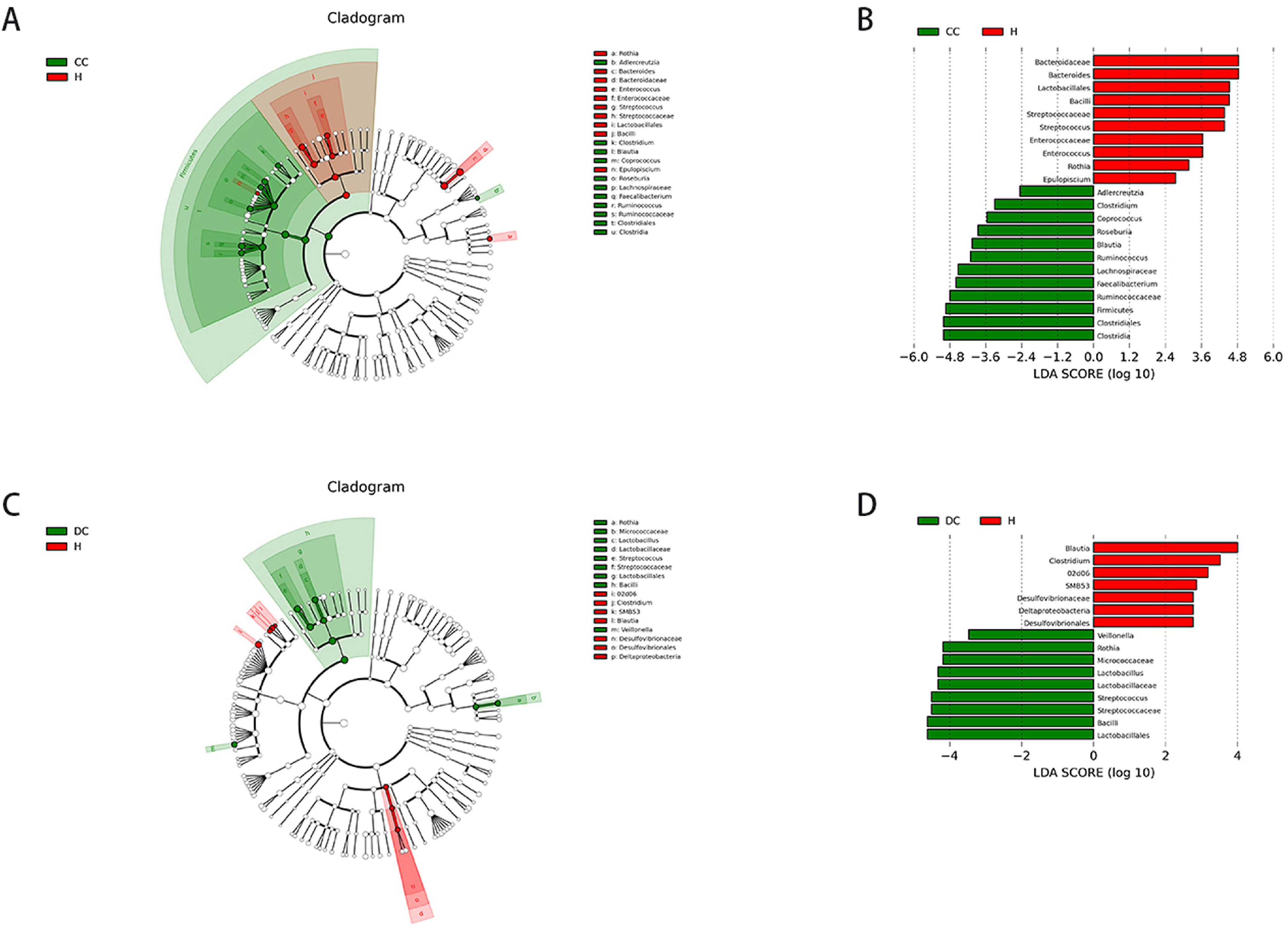
Linear discriminant analysis (LDA) effect size (LEfSe) modelling was applied to identify specific bacterial taxa associated with different stages of cirrhosis (Fig. 4).
 scores for differentially abundant genera between groups (logarithmic LDA score higher than 2 indicated a higher relative abundance in the corresponding group than in the other group).")
Comparison of different groups of microflora differences using the LEfSe online tool. A and C, taxonomic cladogram botained from LFfSe analysis of 16S sequences, taxonomic representation of statistically significant differences between groups. The diameter of each circle was proportional to taxon abundance. B and D, histogram of the linear discriminant analysis (LDA) scores for differentially abundant genera between groups (logarithmic LDA score higher than 2 indicated a higher relative abundance in the corresponding group than in the other group).
There were markedly significant differences in community compositions in HBV-RC patients compared with healthy individuals. In compensated cirrhosis group, there were 22 significantly different taxa. The 5 most abundant genera were Clostridiales (LDA score = 5.01, p = 0.006), Firmicutes (LDA score = 4.94, p = 0.015), Bacteroidaceae (LDA score = 4.84, p = 0.043), Ruminococcaceae (LDA score = 4.80, p = 0.050) and Faecalibacterium (LDA score = 4.59, p = 0.035). In decompensated cirrhosis group, there were as many as 17 significantly different taxa. The 5 most enrichment genera were Lactobacillales (LDA score = 4.63, p = 0.005), Bacilli (LDA score = 4.63, p = 0.008), Streptococcaceae (LDA score = 4.51, p = 0.002), Rothia (LDA score = 4.20, p = 0.009), Blautia (LDA score = 4.00, p = 0.019). These significantly different microbiota can be used as potential biomarkers (Tables S1–S2).
The Firmicutes/Bacteroidetes ratio had been used to evaluate inflammatory related indicators. In our study, the abundance of Bacteroidetes was increased and that of Firmicutes was reduced in patients with HBV-RC compared to the healthy individuals. The above observed results suggested that the higher Firmicutes/Bacteroidetes ratio may accelerate the progression of cirrhosis and inflammation.
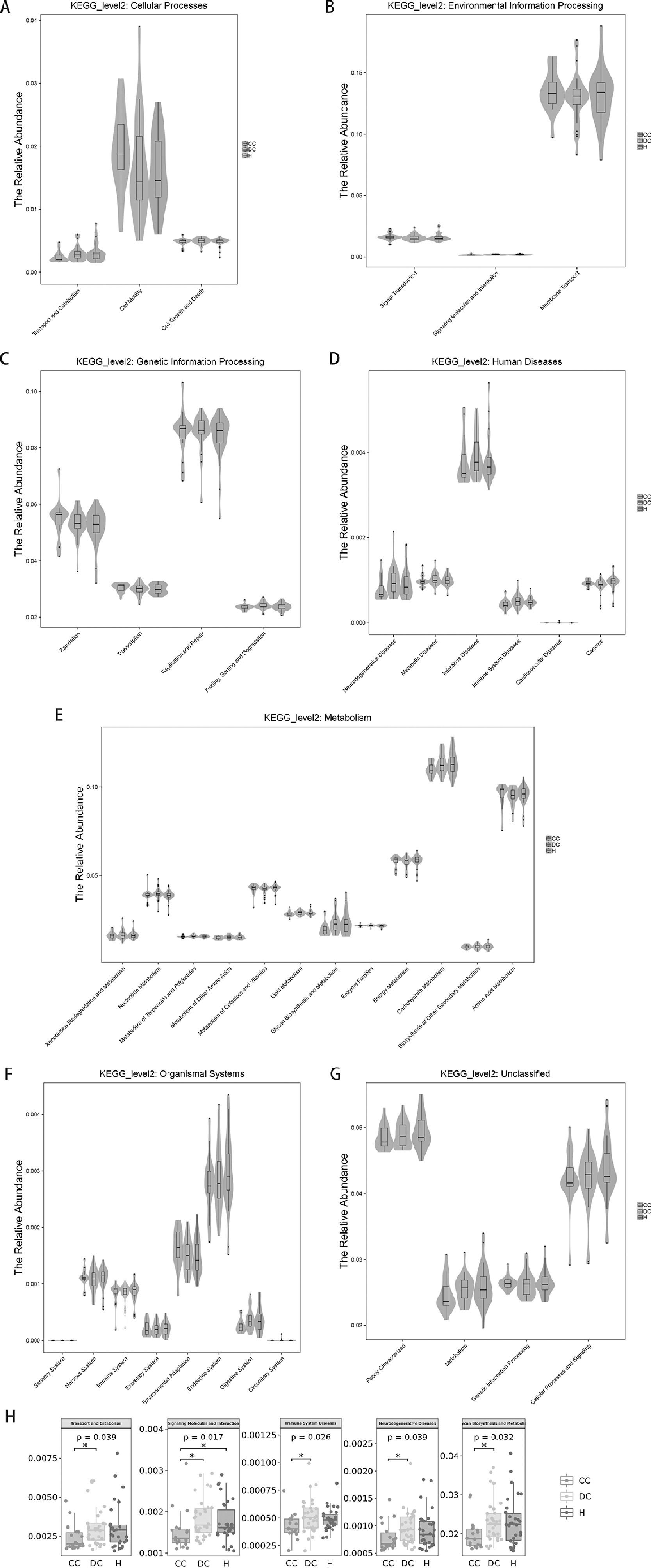
3.8Prediction of microbial metabolic functionThe above analysis focuses on the composition and structure of the microflora. For the study of microbial ecology, the most important concern is undoubtedly the metabolic function of the flora. PICRUSt analysis (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) was performed by comparing 16S rRNA gene sequencing data with a microbial reference genome database. A total of 39 different metabolic functions were predicted in this study. The most enrichment was membrane transport (13.17%), carbohydrate metabolism (11.22%), amino acid metabolism (9.53%), replication and repair (8.50%), energy metabolism (5.78%) and translation (5.33%).
As expected in Fig. 5 regarding the gut microbiota functions associated with human diseases, the subset of genes associated with infectious disease weres significantly enriched in our study population (Fig. 5D). For liver cirrhosis groups, the bacterial functions gene of transport and catabolism, glycan biosynthesis and metabolism were found to be less abundant in those with compensated group than in decompensated group (P = 0.039 and 0.032 respectively).

Prediction of microbial functional composition from 16s rDNA sequencing data with PICRUSt. A–G, representing the differences at the cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, organismal systems and unclassified level among healthy and liver cirrhosis group, respectively. H, representing all statistical differences in microbial metabolic function. P values less than 0.05 are not listed.
For the HBV-RC patients, the distribution densities of signaling molecules and interaction were different. signaling molecules and interaction were higher abundance in HBV-RC patients than in healthy individuals (P = 0.017). Surprisingly, functional predictions regarding neurodegenerative disease and immune system disease related genes were also significantly higher in decompensated group compared with compensated group. Other predicted functional genes including metabolism of terpenoids, transport, catabolism and polyketides translation, etc. also showed differences among different groups (Fig. 5). These observed results suggested that changes in the bacterial composition can significantly alter gene function, which may contribute to the development of cirrhosis.
4DiscussionAlthough many studies have suggested that the pathogenesis of cirrhosis is primarily related to viral and host factors [8], the role of other environmental factors in the progression of HBV-RC, such as the microbiome, remains unclear. In this study, we showed for the first time in-depth integrative analyses of gut microbiota profile of cirrhosis patients. We investigated the gut microbiota in patients with HBV-RC and healthy individuals and our analysis demonstrated a significant difference in microbiota diversity among the HBV-RC and healthy groups.
In our study, analysis of α-diversity using Shannon, Simpson, Chao1 and ACE diversity indexes showed no significant differences in three groups. Nevertheless, some study showed that α-diversity was increased from cirrhosis to healthy controls and positively correlated to Child Pugh scores [9]. The differences may be due to the geographic area, differences in the etiology of the diseases, the target sequencing regions of 16S gene, the platform used, the sequencing depths, or the database selected. In addition, our results suggests that the composition of the gut microbiota had changed in the early stages of cirrhosis. Furthermore, metabolic changes at different stages of cirrhosis were also observed by PICRUSt analysis. These results suggest that changes in the composition of gut microbiota have a potentially pathogenic role in patients with cirrhosis.
Clinically, the cocci to bacilli ratio of the fecal sample is often tested in HBV-RC patients. Most of them showed the imbalance of cocci and bacilli, suggesting intestinal infection or gut microbiota disorder [10,11]. Likewise, in this study, the ratio of cocci to bacilli was significantly elevated in the liver cirrhosis group compared with the healthy group. In terms of taxonomic distributions, the most abundant phyla were Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Fusobacteria and Verrucomicrobia, together comprising 92.1% of the total sequences. We further identified more than 17 genera with different richness in compensated and decompensated cirrhosis groups. The compensated group presented a high relative abundance of Faecalibacterium while the decompensated group showed a high relative abundance of Streptococcus. Further, metagenomic sequencing showed that the richness of Ruminococcaceae was significantly higher in the compensated cirrhosis group than in the decompensated cirrhosis group, while Enterobacteriaceae showed a significantly higher richness in the decompensated group than in the compensated group. Taking together, these findings highlighted an important role for the composition of gut microbiota in the progression of liver cirrhosis which has important clinical implications.
Bifidobacterium, belongs to the gram-positive bacillus of Actinomycetes phylum, is considered one of the most important probiotics, could provide benefits to human health through a variety of mechanisms, including adjust the composition of gut microbiota, reduction in plasma and intestinal endotoxin levels, antibacterial factor production, the improvement of the intestinal barrier function and modulation of local and systemic immunity. [10]. As shown in the study, Bifidobacterium was significantly decreased in the HBV-RC group including compensated cirrhosis patients and decompensated cirrhosis patients, compared with the healthy control. Lactobacillus is one of the most studied and applied probiotic species. Nevertheless, we found that the decompensated cirrhosis group showed a higher relative abundance of Lactobacillus compared with the other groups, suggesting that Lactobacillus was associated with disease progression in liver cirrhosis and seemed contradictory to its probiotic function. As the composition of gut microbiota is influenced by many factors and the environment, the same bacteria may play distinctive roles in different intestinal states. Therefore, the exact role of Lactobacillus in the progression of liver cirrhosis needs further characterization. Additonally, some study found that Lachnospiraceae and Ruminococcaceae participate in carbohydrate fermentation into short-chain fatty acids (SCFAs) in human intestines [12]. Among SCFAs, microbial-derived butyrate regulates the differentiation of Treg cell [13]. In our study, the richness of Lachnospiraceae and Ruminococcaceae were significantly lower in the decompensated cirrhosis group than in the other groups, indicating that the decrease in these bacteria causes a failure in the differentiation of Treg cells, and accelerate the process of decompensated cirrhosis. Summary, the decrease in probiotics, such as Bifidobacterium, Lachnospiraceae and Ruminococcaceae as mentioned above, may disruption of the intestinal mucosal barrier of HBV-RC patients, and thereby promote the development of infection and inflammation.
Based on the composition and structure of the gut microbiota, the metabolic function of the microflora was further analyzed through PICRUSt in this study. We predicted that bacterial functions were mainly enriched in membrane amino acid metabolism, carbohydrate metabolism, transport, repair and replication, translation and energy metabolism. Moreover, gut microbiota dysbiosis is closely related to the occurrence and progression of infectious diseases. Interestingly, we observed that the signaling molecules and interaction were less abundant in patients with HBV-RC than in the healthy cohort. One possible reason is that impaired liver synthesis results in a decrease in the number of transmitters that make signals between cells, and damage to liver tissue in HBV-RC patients can lead to metabolism disorders.
There are still some limitations to this study. Firstly, the differences in patients’ antibiotic used before and after admission, personal alcohol drinking history, may have an impact on the results of the microbiota. Secondly, our study does not elucidate the direct influence of microbiota on the progression of liver cirrhosis. In future studies, germ-free animals and additional bio functional analyses are needed to further unconver the causal relationship between gut microbiota dysbiosis and HBV-RC.
In conclusion, our study demonstrated that the composition of gut microbiota changed at different phases of HBV-RC. Changes in gut microbiota composition could be a biological factor of progression of liver cirrhosis. We believe the gut microbiome in HBV-RC patients may provide a useful prognosis marker for disease progression, outcomes prediction and treatment, and further microbiome-based studies may provide new insights into the pathogenesis of HBV-RC, the etiology of its progression and the novel therapeutic strategy such as the use of probiotics or fecal microbiota transplantation.
The study was supported by grant from Startup Fund for scientific research, Fujian Medical University [2019QH1101]. We also thank the generous volunteer subjects who enrolled in the study.




{kind=link}
{kind=link}