




Background/Aims: Orthotopic liver transplantation is being used with more frequency as the treatment for Wilson's disease. The experience at the Instituto Nacional de Ciencias Medicas y Nutricion Salvador Zubiran with orthotopic liver transplantation for Wilson's disease is reported. We perform an extensive literature review for this treatment modality.
Methods: Between january 2000 and june 2003, 23 orthotopic liver transplants were performed at our institution, 2 of them for Wilson's disease. Both the patients presented with chronic advanced liver disease and one presented neurologic dysfunction.
Results: Both the patients were trasplanted without any major complication and are alive 43 and 22 months after the transplant respectively. To our knowledge 370 liver transplants have been reported in the international literature since 1994 for the treatment of Wilson's disease.
Conclusions: Currently, orthotopic liver transplantation should be considered as a major option for the treatment of chronic liver disease in patients with Wilson's disease. Although it is well known that the transplant only partially corrects the defective metabolism in patients with Wilson's disease, it does convert the copper kinetics of a homozygous to that of a heterozigote, thus, providing an effective phenotypic cure.
Wilson's Disease (WD), Orthotopic liver transplantation (OLTx)
IntroductionSamuel Alexander Kinnier Wilson (1878-1937) while being a resident in London's National Hospital saw 4 young patients with cirrhosis whom he observed were all suffering from the same fatal mysterious familial neurologic disorder, and in 3 of whom he performed the autopsy and neuropathologic examination himself. Wilson discovered that the syndrome had actually been reported approximately 20 to 50 years earlier in 9 other cases, although the significance of the malady and, especially, the importance of cirrhosis were not previously appreciated. Wilson fastidiously characterized a neurologic syndrome of tremors and involuntary movements (chorea and athetosis), muscular rigidity and contracture, dysarthria and, often, emotional lability. He correlated the clinical findings with degeneration of the lenticular nucleus, involving cavitation of the putamina and less severe degeneration of the globus pallidus; hence, Wilson's term of progressive lenticular degeneration for this newly recognized disorder. The theoretic importance of Wilson's observation was that it was the first study of the anatomy, function and disorder of the "extrapyramidal system" - a name he coined, in which a link was made between a movement disorder, degeneration of the basal ganglia and cirrhosis of the liver.2
Wilson saw that cirrhosis was integral to the syndrome of progresive lenticular degeneration and even postulated that the morbid agent is probably a toxin associated with the hepatic cirrhosis.
Two years after Wilson's publication, the finding of excess hepatic copper in what was still called pseudosclerosis was seemingly insufficient to incriminate it as the toxin (silver was erroneously blamed then), which took another 35 years until Cumming showed that all patients dying of Wilson disease (WD) had excess copper in both liver and brain (especially in the lenticular nucleus). Copper deposition is found in many other tissues of the body, including the characteristic corneal rings (described by Kayser and later Fleischer).2,11-13
An apparent early breakthrough in undestanding the molecular bases of WD was the paradoxical finding of low ceruloplasmin concentrations in the serum of patients with the disorder. This approach was explored because it was known that there was excess copper in WD patients, and ceruloplasmin is the major copper-containing protein in the circulation. This deficiency is a consequence and not a cause of WD, linkage analysis and restriction fraction polymorphism mapped the putative abnormal gene to the q14-q21 band of chromosome 13. The gene itself was sequenced by 3 separate groups independetly and shown to code for a copper-transporting P-type ATPase, ATP7B, for which the intracellular location and precise biological function were initally enigmatic, especially its role in the biliary excretion of copper that is characteristacally impaired in WD.
Today, WD is described as an heritable disorder of copper metabolism characterized by an inability of the liver to excrete the metal into the bile. As a result of this defect in biliary excretion of copper, the hepatic content of this metal increases and leads to progresive hepatocellular injury and ultimately cirrhosis. As the liver becomes progressively saturated with copper, excess copper spills over into other tissues and the copper content of the basal ganglia, cornea, bones, kidneys increases, producing pathophyiologic consequences characteristic of WD.1,8,13
Although several medical therapies exist for the control of WD, none cures the disease. The institution of specific medical therapy allows for stabilization of the disease and frequently functional recuperation, often even to normality as long as the disease process has not progressed to far. In advances cases, little or no recovery occurs with medical therapy, although there is a suggestion of prolonged survival most patients with Wilson have chronic active hepatitis and cirrhosis. However, all of the currently available medical therapies have consistently failed to prevent death associated with a fulminant presentation of WD and in those that have far advanced chronic, irreversible liver disease, many of whom also have advanced neurologic complications at the time of initial diagnosis. As a result, orthotopic liver transplantation (OLTx) has increasingly been used to treat WD with these problems during the last decade. Although the first successful OLTx for WD was performed in 1969, and there are recent reports, still, there is little information in the medical literature about the efficacy of OLTx for WD.1,8
The pourpose of this study is to present the experience at the Instituto Nacional de Ciencias Medicas y Nutricion Salvador Zubiran with OLTx for WD, along with an extensive review of the literature for this treatment modality.
ResultsSince January 2000, 23 OLTx have been performed at our institution for different advanced irreversible liver disease (Table I) including 2 cases of WD. Both the patients were male 17 y/o and 19 y/o respectively.
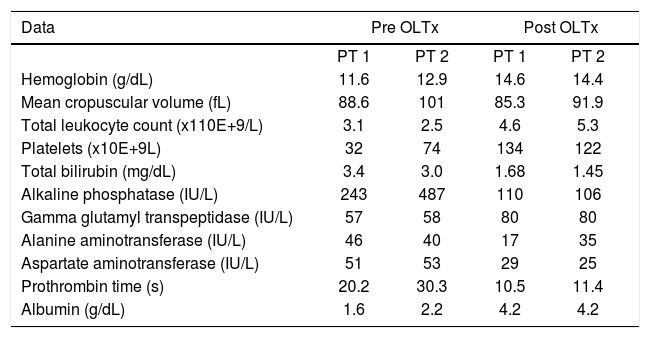
Case reportWe have performed two OLTx for WD at our institute since 2000. In both cases the diagnosis of WD was based upon a combination of the presence of Kayser-Fleischer rings, a low serum ceruloplasmin level, elevated 24-h urine copper excretion and a quantitative hepatic copper measurement. Both the degree of disability and overall performance of each patient were noted in a pre and postransplant fashion. The critical laboratory data are listed in table II.
Critical Laboratory data.
| Data | Pre OLTx | Post OLTx | ||
|---|---|---|---|---|
| PT 1 | PT 2 | PT 1 | PT 2 | |
| Hemoglobin (g/dL) | 11.6 | 12.9 | 14.6 | 14.4 |
| Mean cropuscular volume (fL) | 88.6 | 101 | 85.3 | 91.9 |
| Total leukocyte count (x110E+9/L) | 3.1 | 2.5 | 4.6 | 5.3 |
| Platelets (x10E+9L) | 32 | 74 | 134 | 122 |
| Total bilirubin (mg/dL) | 3.4 | 3.0 | 1.68 | 1.45 |
| Alkaline phosphatase (IU/L) | 243 | 487 | 110 | 106 |
| Gamma glutamyl transpeptidase (IU/L) | 57 | 58 | 80 | 80 |
| Alanine aminotransferase (IU/L) | 46 | 40 | 17 | 35 |
| Aspartate aminotransferase (IU/L) | 51 | 53 | 29 | 25 |
| Prothrombin time (s) | 20.2 | 30.3 | 10.5 | 11.4 |
| Albumin (g/dL) | 1.6 | 2.2 | 4.2 | 4.2 |
The first patient was 17 y/o when we first saw him. He presented with jaundice, fever, generalized weakness and signs of hypogonadism and was first diagnosed with HAV in another institution. He arrived at our institution ER with involuntary left lower extremity movements of about 2 weeks of evolution. After studying his case, revealing a low cerulplasmin level of 8 mg/dL (22-55 mg/ dL)), and a urinary copper excretion of 967 mcg (<60 mcg/24h) and Kayser-Fleischer rings in the ophtalmoscopic examination. A diagnosis of Wilson disease was made. The patient had a history of SBP in 1997. The patient Child - Pugh score at the moment was C. An upper endoscopy study revealed congestive gastrpathy. The abdominal US showed chronic hepatopathy and ascitis. A head MRI showed hyperdensity in the basal ganglia predominantely in the caudate and lenticular nucleus. 7 months after he was listed in the hepatic transplant waiting list he received a liver transplant without major complications. He is alive after 43 months of follow up.
The second patient presented with complaints of cutaneous hyperpigmentation, hepatic (Child-Pugh score C) and pancreatic function alterations and data of hypogonadism, He also showed pericorneal pigment compatible with Kayser-Fleischer ring, he had a serum ceruloplasmin level of 4 mg/dL (22-55 mg/dL) and a urinary copper excretion of 238 mcg (<60 mcg/24h) establishing the diagnosis of WD. An upper endoscopy showed esophageal varices grade III-IV, with data sugestive of poor prognosis, like varices on top of other varices and congestive erosive gastropathy. The supra-hepatic gradient pressures measured revealed sinusoidal portal hypertension. The hepatic biopsy was compatible with chronic hepatopathy, fibrosis and material deposits sugestive of copper or hemosiderin. The abdominal US revealed chronic hepatopathy, splenomegaly and ascitis. The head MRI demonstrated material deposits in the caudate nucleus, and basal ganglia. 14 months after he was diagnosed he received a liver transplant without any major complication, he is alive 22 months after the transplant.
Both patients maintain a normal daily routine, being both college students, participating in all academic and extracurricular activities.
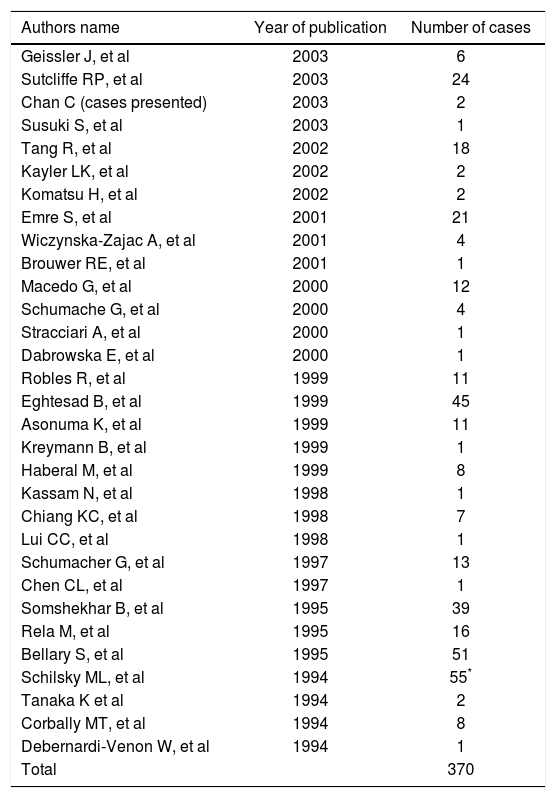
DiscussionAccording to the data presented, OLTx should be offered to patients with WD who present with either a fulminant/subfulminant course or more typically with chronic advanced end stage liver disease and many of its serious complications. The data from our experience and that reviewed in the literature demonstrates that liver transplantation partially corrects the underlying metabolic defect of WD to a level found in disease free heterozigotes as assessed by formal copper kinetic studies. To our knowledge 370 cases have been reported in the literature since 1994 (Table III). Although liver transplantation does not provide a perfect cure (it does not produce a plasma radioactive copper response consistent with an unaffected individual), it does produce an effective cure by converting the response to one similar enough to that found in an obligate heterozigote for WD.1,3,4,7-9 The reasons why liver transplantation does not fully correct the copper kinetic measurements to a normal level is still unknown. Possible explanations are a) the immunosupression used alters the patterns of copper metabolism to a pattern similar to that seen in heterozigotes; or b) that a factor originating from a non hepatic source in an individual with genetic WD continues to moderate hepatic copper metabolism in a manner suggestive of a genetic heterozigote for WD. Somewhat similar data have been found for individuals transplanted for other liver based metabolic disorders, such as tyrosinemia, alpha-1-antitrypsin deficiency, type II hyperlipoproteinemia and type II hyperoxaluria. In all of this cases the consequences of the primary metabolic disease are corrected by OLTx but still, a subtle evidence of the residual primary disease expression persists in the liver transplant recipient. It is of the most importance to note that the heterozygote state for each of these metabolic abnormalities is not known to be associated with clinical disease, and thus liver transplantation effectively produces a clinical cure, despite the failure to achieve complete metabolic normalization. The single fact that liver transplantation cures WD is reflected in the total normalization of serum copper, ceruloplasmin and liver chemistry levels following an OLTx.
OLTx for WD reported in the literature since 1994.
| Authors name | Year of publication | Number of cases |
|---|---|---|
| Geissler J, et al | 2003 | 6 |
| Sutcliffe RP, et al | 2003 | 24 |
| Chan C (cases presented) | 2003 | 2 |
| Susuki S, et al | 2003 | 1 |
| Tang R, et al | 2002 | 18 |
| Kayler LK, et al | 2002 | 2 |
| Komatsu H, et al | 2002 | 2 |
| Emre S, et al | 2001 | 21 |
| Wiczynska-Zajac A, et al | 2001 | 4 |
| Brouwer RE, et al | 2001 | 1 |
| Macedo G, et al | 2000 | 12 |
| Schumache G, et al | 2000 | 4 |
| Stracciari A, et al | 2000 | 1 |
| Dabrowska E, et al | 2000 | 1 |
| Robles R, et al | 1999 | 11 |
| Eghtesad B, et al | 1999 | 45 |
| Asonuma K, et al | 1999 | 11 |
| Kreymann B, et al | 1999 | 1 |
| Haberal M, et al | 1999 | 8 |
| Kassam N, et al | 1998 | 1 |
| Chiang KC, et al | 1998 | 7 |
| Lui CC, et al | 1998 | 1 |
| Schumacher G, et al | 1997 | 13 |
| Chen CL, et al | 1997 | 1 |
| Somshekhar B, et al | 1995 | 39 |
| Rela M, et al | 1995 | 16 |
| Bellary S, et al | 1995 | 51 |
| Schilsky ML, et al | 1994 | 55* |
| Tanaka K et al | 1994 | 2 |
| Corbally MT, et al | 1994 | 8 |
| Debernardi-Venon W, et al | 1994 | 1 |
| Total | 370 |
For many authors OLTx is indicated for WD when there is acute hepatic failure and severe hepatic insufficiency not responsive to chelation treatment. Indications for OLTx in patients with progressive neurologic deterioration without hepatic insufficiency, who do not improve with correct medical treatment is still widely debated.5,10



