




Introduction and aim. Autophagy and its regulated pathways participate in many important cellular physiology and pathological processes involving protein aggregates, damaged mitochondria, excessive peroxisomes, ribosomes, and invading pathogens. This study aimed to review recently published studies and further describe the long noncoding RNA (lncRNA)-regulated autophagy during drug-induced liver injury (DILI).
Material and methods. DILI, autophagy, autophagy-related genes (ATGs), and lncRNA were used as key words to search published studies from PubMed, Google Scholar, and Web of Science. All related studies were reviewed and analyzed.
Results. Many studies explicitly indicated that DILI and its progression to acute liver failure were causatively linked to endoplasmic reticulum stress and subsequently induced autophagy, which protect hepatocytes during DILI. LncRNA, as a noncoding RNA, influences the regulation of the expression of ATGs to manipulate autophagy.
Conclusions. This review described the recent findings on autophagy and its possible lncRNA-miRNA-associated pathways, thereby providing new insights for further studies on the pathogenesis of DILI.
Drug-induced liver injury (DILI) has gained significant attention worldwide in the last decades on account of its significant morbidity and mortality.1–4 It is one of the most common causes of acute liver failure to date in the United States and Europe, and a leading reason for emergent liver transplant.5,6 Further understanding of the pathogenesis of DILI might have important value for preventing and treating the disease. The pathogenesis of drug-induced hepatitis is considerably complex, involving multiple pathological processes, such as liver cell damage, intracellular fat change, fibrosis, apoptosis, necrosis, and so on.7 Although the pattern of DILI can be generalized as intrinsic types referring to drugs with dose-dependent and idiosyncratic types characterizing by an adaptive immune attack,8 the common downstream events were the imbalance between pathways to injury and restorative tissue repair.9 It is related to not only the type and nature of the drug, but also the genetic background of the individual. However, the ultimate common pathway is the activation of the cell death signal.10,11 Results from a genomics approach revealed that endoplasmic reticulum (ER) stress and nuclear factor-erythroid 2-related factor 2 (Nrf2)signaling are two major pathways involved in DILI. Other is subsequent protein kinase R-like ER kinase (PERK), activating transcription factor 4 (ATF4), and C/EBP homologous protein (CHOP) activation, which may be the crosstalk with pro-apoptosis signaling.12 It is supported by the research on CHOP knockout (KO) mice that can protect liver cells from APAP-induced damage and reduce necrosis, exhibiting proliferation and increased hepatocyte survival.13 Therefore, ER stress, which has dual effects either cause apoptosis or protective effect, is the key process in the pathogenesis of DILI. In neurons, ATF5 upregulation induced mainly by ER stress does not lead to apoptosis, implicating that ER stress acts as a prosurvival mechanism.14 Moreover, inhibition of IRE1a followed by ER stress enhances the cytotoxic effects.15 The role of ER stress in promoting cell survival and death is still unknown. Therefore, this review focused on the molecular mechanism of autophagy referring to restorative tissue repair regulated by long noncoding RNA (lncRNA) for a better understanding of the pathogenesis, diagnosis, and treatment of DILI.
Autophagy and DiliAutophagy is a process designed to sequester and degrade intracellular components, beginning with the formation of double-membrane vesicles tightly controlled by the autophagy-related genes (ATG). It is a critical adaptive response for cell survival, which can be dramatically induced by starvation and a broad range of stressors, such as ER stress, pathogen infection, oxidative stress, and so on.16 The failure to get rid of intracellular wastes by autophagy greatly disrupts homeostasis, which can invariably induce cell death and inflammation consequently resulting in tissue injury. Autophagy acts as an intracellular degradation process associated with lysosomes. It mainly targets cy-tosolic components such as abnormal proteins and or-ganelles.17 It is essentially a self-repairing process to deal with potential risk factors for intracellular physiological processes and prolong the life of cells via energy regeneration18,19 and removal of harmful or useless substances. Otherwise, apoptosis can sequentially be elicited as another protective factor to relieve stress when autophagy fails to perform its functions.20 So far, more than 40 autophagy-related genes have been identified in yeast and mammals, which tightly control the intracellular adaptive process via multiple signaling pathways.21 A total of 15 core ATG genes (ATG1-10, 12-14, 16, and 18), which are highly conserved in mammals, are required for selective autophagy for protein aggregates, damaged mitochondria, excessive peroxisomes, ribosomes, and invading pathogens. The expression and regulation of these genes are closely related to the death pathway.21,22
The hepatocytes have a higher capacity for autophagy compared with other cell types because of a large number of ER, mitochondria, and lysosomal enzymes required for multiple metabolic functions and to cope with different biological or chemical stimuli, ultimately ensuring cellular homeostasis.23,24 The ER is one of the main sites for drug metabolism in the liver, and also the assembly and modification of secretory and integral membrane proteins. ER stress is the stimulus for autophagy. Further, ER is the initiation site for the biogenesis of autophago-somes.22,23 ER stress and autophagy are related to apoptosis and hence have been recognized as critical pathways in the regulation of cell death.24,25 Recent studies have demonstrated essential protective functions of autophagy in hepatocytes during liver injury.15,26–34 Thymidine analogs were found to inhibit autophagy on constitutive and induced levels in hepatocytes, increasing reactive oxygen species production, lipid accumulation, and hepatic dysfunction, and thereby resulting in apoptosis.28 Doxorubicin, an antibiotic drug used for chemotherapy in various cancers, induces cardiotoxicity, which is consistent with dysregulated autophagy and autophagosome formation shown by a variety of approaches in cardiomyocytes.30 Knockout of ATG5 the autophagy gene has confirmed that deficiency of autophagy can increase acute liver injury induced by D-galactosamine/lipopolysaccharides in mice. Similar results were observed with a knockout of the gene Atg7, leading to liver injury with a hepatocyte-specific loss of autophagy.32,35 Deletion of the autophagy gene Atg7 led to the accumulation of concentric membranous structures, which were continuous with the rough ER. These structures were surrounded by various aberrant mitochondria and lipid droplets. Disorganized hepatic lobules and cell swelling were noted on histological analysis, while no hepatocyte proliferation or regeneration was observed. These findings were consistent with the protective role of autophagy in eliminating abnormal organelles for maintaining normal liver metabolism.36 ER is an original site for autophagy. Moreover, autophagy closely related to ER is essential in protecting the hepatocytes from damage in metabolic processes.
Drug metabolism in hepatocytes primarily occurs on ER. Hence, accumulation of reactive metabolites and conjugate proteins can strikingly increase the ER stress in response to overdose or long-term use of drugs.37–39 ER stress is resolved by the activation of the unfolded protein response, which can be elicited by three stress sensors inositol-requiring protein 1α, activating transcription factor 6, and protein kinase RNA-like ER kinase to orchestrate the normal ER function.40 The collaboration of these processes helps in deciding the scale of adaptive capacity and thus governs whether cells will re-establish homeostasis or activate cell death programs via an internal pathway of apoptosis with inhibition of autophagy.41 Existing data implicated that autophagy, as a downstream mediator of ER stress, had a pronounced decrease in cell viability during drug-induced damage of rat hepatocytes.42 The mechanism underlying hepatotoxicity caused by drugs has not been recognized yet. However, it seems that these mechanisms are closely related to the increase in damaged organelles and nonfunctional proteins during biotransformation to metabolites, resulting in increasing ER stress and oxidative stress. For instance, acetaminophen (APAP), which is widely used as an antipyretic and analgesic drug, is the most common source of severe hepatotoxicity. APAP-induced hepatotoxicity has been demonstrated to be mediated mainly via its reactive metabolite. During APAP overdose, N-acetyl-p-benzo-quinone imine (NAPQI), which is generated from APAP metabolism by cytochrome P450, can deplete hepatic stores of glutathione, interfering with cellular redox homeostasis.43,44 The remaining NAPQI reacts with many cellular proteins and induces autophagy acting as an adaptive response to remove damaged organelles, especially mitochondria. Furthermore, induction of autophagy inhibited APAP-induced hepatotoxicity, preventing against liver injury.45,46 In addition to ER stress, NRF2-mediated oxidative stress response has implicated in DILI, which suggested excessive reactive oxygen species (ROS) were generated by the oxidative metabolism of drugs.47 It is generally believed that ROS result from both the NAD(P)H oxidase (NOX) and CYP450 system using co-factor NADPH, which is following by the oxidative damage mainly manifested as lipid peroxidation.48 However, ROS generation can be largely attributed to a NOX activity rather than the CYP450 system when incubated acetaminophen and NADPH in microsomes of rat liver.49 The recent development indicated that autophagy-deficient caused oxidative stress increasing in melanocytes, also the upregulation of ROS, Nrf2 antioxidant signaling and lipid oxidation.50 Whether these phenomena existing in hepato-cytes need further experiments to prove. Although the hepatocyte damage induced by different drugs may have varied pathways, the central factors are the reactive metabolites, resulting in abnormal proteins or organelles interfering with the normal physiological program. This provides the framework for thinking about ER stress-induced autophagy, which is a cellular adaptive response to cope with these stresses involved in liver injury caused by drugs. Researchers suggested ER stress and au-tophagy can constitute an effective defense mechanism against multiple insult from lipid metabolism, alcohol and non-alcoholic fatty liver in hepatocytes which may be similar to DILI.51 It has been proven autophagy-enhancing drugs can alleviate liver steatosis, liver injury, and dyslip-idemia in both alcohol-fed and HFD(high fat diet)-fed mice,52 yet it is not clear how autophagy is initiated in these contexts that may be different from DILI. Further studies to clarify the intensity of autophagy induced by ER stress, particularly originating from drug biotransformation in hepatocytes, might provide important information on DILI.
Regulation of AutophagyThe performance of autophagy includes two steps: formation of autophagosomes and generation of autolyso-somes from the fusion of double-membrane vesicles with lysosomes.53 The autophagosomal membrane originates from the ER,54 as confirmed by two groups that demon-strated the physical connection between the ER and isolation membrane using 3D electron microscopy.55,56
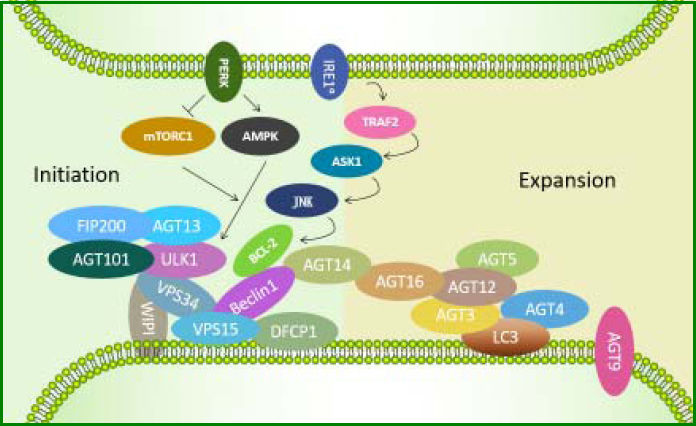
So far, three critical protein complexes have been identified, including the ULK1-ATG13-FIP200-ATG101 complex, the Beclin1-ATG14-Vps34-Vps15 (class III PI3-kinase) complex, and the ATG12-ATG5-ATG16L1 complex. The ULK1 complex appears to be the most upstream unit, followed by ATG14-containing PI3-kinase complex, which together regulate the formation of both DFCP1 and WIPI-1 structures. Most downstream events are activation of the ATG12 system and the LC357 (Figure 1). These proteins participate in different steps of autophagy in a temporal order based on their roles in autophagosome formation and are regulated by each other through diverse pathways relying on different intracellular stimuli or different cell types.58 Therefore, regulation of autophagy involves not only the interaction between the core ATG proteins but also variation in ATG genes at the transcrip-tional and posttranslational levels.50 Autophagy occurs at a constitutive basal level but can be upregulated by different types of stresses such as starvation, damaged intracellular components, or pathogen infection.59 It first involves the recruitment and co-working of core ATG proteins,60 followed by changes at the transcriptional and post-tran-scriptional levels,58 which probably govern the extent of autophagy to cope with a strong or prolonged stimulus.
 stress. First, the ULK1-ATG13-FIP200-ATG101 complex is activated. Then, PtdIns3K class III complexes (Beclin1-VPS15-VPS34) are induced to transform phosphatidylinositol 3-phosphate (PtdIns3P) intensified by ATG14, followed by recruitment of downstream effectors such as DFCP1 and WIPI-family. ATG12-ATG5-ATG16L1 complex exerts its E3-like function and thus catalyzes LC3-I, leading to LC3 cleavage by ATG4 to LC3-II.")
ATGs involved in the initiation of autophagy induced by endoplasmic reticulum (ER) stress. First, the ULK1-ATG13-FIP200-ATG101 complex is activated. Then, PtdIns3K class III complexes (Beclin1-VPS15-VPS34) are induced to transform phosphatidylinositol 3-phosphate (PtdIns3P) intensified by ATG14, followed by recruitment of downstream effectors such as DFCP1 and WIPI-family. ATG12-ATG5-ATG16L1 complex exerts its E3-like function and thus catalyzes LC3-I, leading to LC3 cleavage by ATG4 to LC3-II.
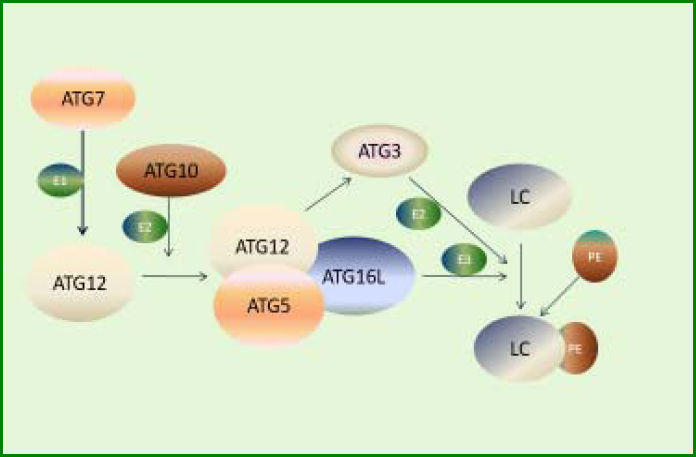
Recent progresses in autophagy research in genetically modified mice (especially the study on Atg5 KO mice) have revealed the important role of some ATG genes, indicating the importance of autophagy for survival.61 Other four phenotypes of Atg7, Atg9, and Atg16L1 have shown a result similar to that in Atg5 using the gene KO mice model. Atg5, Atg7, Atg9, Atg16L1, Beclin 1, and FIP200 are indispensable components for the formation of autophagosomes,62 implying the involvement of these proteins in the regulation of autophagy. In line with these findings, the expression of ATG3, ATG5, ATG12, ULK1, LC3B, Beclin-1, and GABA(A)receptor-associated protein like 1 was downregulated in human osteoarthritis cartilage compared with the normal cartilage, whereas the expression of mTOR was significantly upregulated. The expression levels of these proteins could be subsequently restored on deleting mTOR gene in the cartilage of mice.63 The involvement of ATG5, ATG7, ATG9, and ATG16L in the conjugation system, which directly participates in the formation of phosphatidylethanolamine with the expansion of autophagosomes (Figure 2),60 suggests that the intermediate stage presumably is vital in regulating the extent of autophagy. For instance, an inhibitor of soluble epoxide hydrolase can recover the levels of Atg12-Atg5 and LC3-II conjugates, resulting in the upregulation of hepatic autophagy to modulate inflammation in fat-1 mice.64 Also, deletion of Atg5 in hepatocytes can lead to loss of au tophagy, consequently increasing apoptosis, inflammation, and fibrosis.65 During prolonged starvation, the expression level of SQSTM1/p62, a substrate protein degraded by autophagy, is determined by three factors: reduction in au-tophagic degradation, activation of transcriptional factors, and availability of amino acids needed to synthesize new proteins.63

ATG12-ATG5 system involved in the expansion of autophagosomes. First, ATG7 activates ATG12 by functioning as an E1-like enzyme. Then, ATG12 conjugates to ATG5 and ATG16L, forming a functional complex as an E3-like enzyme. Meanwhile, activated ATG12 recruits ATG3, which acts as an E2-like enzyme. Finally, LC3 conjugates to PE under the action of the complex and ATG3.
Other substrates involved in autophagy might have similar regulatory processes. A study on the transcriptional regulation of autophagy revealed that the fed-state sensing nuclear receptor inhibited autophagy by binding to 178 (of 230) autophagy-related genes for decreasing their expression, and CREB (the fasting transcriptional activator) promoted autophagic degradation of lipids via upregulating 112 genes under nutrient-deprived conditions in mouse liver.66
These signaling pathways and transcriptional or post-transcriptional factors manipulate autophagy and serve as critical components to dispose of stress or changes in the liver.24
Ample evidence indicates different classes of RNA, ranging from small to long noncoding RNAs, as key regulators of gene expression.67 In a hepatocyte dedifferentiation study, massive alterations of noncoding transcriptome were found, hallmarked by increased expression of small nucleolar RNAs, lncRNAs, microRNAs (miRNAs), and ribosomal genes, preceding changes in protein-coding genes. Notably, only the regulation of lncRNAs among the ncRNAs displayed significant temporal and directional congruity with the profiles of coding genes. This showed that lncRNA primarily took part in regulating the expression of coding genes, influencing protein output. On the contrary, lncRNA might be generated from the transcription of protein-coding genes.68 Above all, it is reasonable to speculate that the regulation of the transcription of ATG was partially manipulated by lncRNA. Therefore, an in-depth understanding of the molecular mechanisms underlying the manipulation of autophagy is crucial to obtain new insights on the strategies of diagnosing and treating autophagy-associated liver diseases.
IncRNA-regulated autophagyLncRNA (length > 200 bases) have attracted the attention of many researchers recently because of their involvement in diverse fundamental processes, in particular gene expression, which refers to recruiting chromatin-modify-ing complexes, enhancing transcription, decoying miR-NAs, providing a scaffold, and encoding peptides.69,70
A study on the evolutionary history of lncRNA revealed that lncRNA promoters were more frequently associated with transcription factors compared with random intergenic regions, suggesting their potential role in tran-scription.71 MiRNA has always been considered as the central player in controlling gene expression at the post-transcriptional level.72 It has been extensively studied as a repressor of gene expression or cleavage of mRNA.18,51,73 During the last decade, increasing evidence has indicated that lncRNA with low or no protein-coding potential may be equally important in the regulation of gene expression.74 A large number of studies have confirmed the role of lncRNA in diverse biological functions via different mechanisms, including regulating the expression of adjoining protein,75 negatively regulating RNA polymerase II (RNAP II),76 and regulating alternative splicing77 and microRNA sponges termed as competing endogenous RNAs (ceRNAs).78 The most important function of RNA is to synthesize the proteins necessary for life activities. The interaction between different kinds of RNA, in particular ncRNA and mRNA, has a decisive role in regulating transcription and translation. After all, communication between the same kind of RNAs takes precedence over heterogeneous crosstalk. The challenge was that these experiments revealed what happened, but not the coordination in time and space. The present study paid close attention to some of the recent data concerning the involvement of lncRNA in regulating different steps of autophagy especially the expression of related proteins, giving a better understanding of the process.
Some studies found that lncRNA APF (AK079427, 1695 nt long) regulates the occurrence of autophagy via inhibiting miR-188-3p in a sequence-specific manner to significantly protect the expression of ATG7 in the myocardial cells of mice.79 In addition, the overexpression of lncRNA (FLJ11812, located in the 3' UTR of TGFB2 gene, contains 1370 nt) could promote autophagy by increasing the ATG13 protein level. Furthermore, FLJ11812 competes to bind with MIR4459, which targets ATG13 to down regulate its expression, which is analogous to the mechanism of APF acting as a competing endogenous RNA (ceRNA) to control ATG13 level80. LncRNA PTENP1 indirectly contributes to autophagy by effectively upregulating ULK1, ATG7, and p62 levels via the antagonizing action of miR-17 in HCC cells.81 Another study confirmed that the overexpression of HOTAIR, an lncRNA that could promote the activation of autophagy in HCC cell lines, was equivalent to upregulating the expression levels of ATG3 and ATG7 (Table 1).82 These findings further explained the importance of the expression of the aforementioned key genes in modulating autophagy. Also, lncRNA had a strong influence on the protein expression. LncRNA has been proved to be crucial in regulating autophagy and participate in the pathophysiological processes of various human diseases, such as NBR2 and tumor development,83 MEG3 and bacterial infections,84 TGFB2-OT1 and in-flammation,85 Risa and insulin sensitivity,86 PCGEM1 and osteoarthritic synoviocytes,87 and so on. The regulation network of gene expression involves lncRNA in au-tophagy, providing a new insight into the mechanism of hepatocyte injury. The liver is found to be rich in lncR-NA, followed by testes and neural tissues in the body.71 Therefore, further understanding of the role of lncRNA during pathophysiological conditions can help in developing potential diagnostic and therapeutic methods for patients with DILI.
IncRNA-miRNA associated with autophagy.
| lncRNA | Targest | Role in autophagy | Diseases | References |
|---|---|---|---|---|
| MALAT1 | miR-30a miR-101 miR-216b miR-124 miR-23b-3p | MALAT1 activates autophagy serving as a molecular sponge for miR-30a that suppresses Beclin1-dependent autophagy and miR-101, RAB5A, and ATG4D It also binds to miR-216b, modulating autophagy It promotes autophagy by directly targeting miR-124 It induces autophagy for acting as a ceRNA for miR-23b-3p to increase the expression of ATG12 | Ischemic stroke; glioma; hepatocellular carcinoma; retinoblastoma; gastric cancer | 88, 89, 90, 91, 92 |
| TGFB2-OT1 | miR-3960 | TGFB2-OT1 acts as a competing endogenous RNA regulating the expression of | Vascular endothelial cell | 85 |
| (FLJ11812) | miR-4488 miR-4459 | the miRNA targets CERS1, NAT8L, and LARP1, which participate in autophagy | dysfunction | |
| APF | miR-188-3p | APF regulates the expression of ATG7 through miR-188-3p | 93 | |
| HULC | miR-6886-3p miR-6825-5p, miR-6845-5p | HULC downregulates miR-6825-5p, miR-6845-5p, and miR-6886-3p to protect autophagy | Hepatocellular carcinoma (HCC) | 94 |
| AC023115.3 | miR-26a | AC023115.3 acts as a competing endogenous RNA for miR-26a, which has an inhibitory effect on GSK3â leading to a decrease in autophagy | Glioma | 95 |
| HNF1A-AS1 | miR-30b | HNF1A-AS1 promotes autophagy by acting as a ceRNA for miR-30b, which can regulate the expression of Bcl-2 and ATG5 | Hepatocellular carcinoma (HCC) | 96 |
| PTENP1 | miR-17 miR-19b miR-20a | PTENP1 induces autophagy through decoying miR-17, miR-19b, and miR-20a, which target autophagy genes as ULK1, ATG7, and p62 | Hepatocellular carcinoma (HCC) | 81 |
| PVT1 | miR-186 | PVT1 induces autophagy by targeting miR-186, which decreases the expression of Atg7 and Beclin1 | Glioma | 97 |
| HOTAIR | miR-454-3p | HOTAIR increases autophagy by inducing DNA methylation of miR-454-3p, which targets STAT3 and ATG12 | Chondrosarcoma | 98 |
| GAS5 | miR-21 | GAS5 suppresses autophagy for acting as a negative regulator of miR-21 | Osteoarthritis | 99 |
| PCGEM1 | miR-770 | PCGEM1 induces autophagy by acting as a sponge for miR-770 | Osteoarthritis | 100 |
Autophagy is the most basic protective pattern for normal physical activities and survival. Exploring the regulatory mechanism influencing the extent of autophagy at the molecular level, especially for protein transcription and translation, can substantially contribute to understanding the pathophysiology of cellular damage. While manipulating autophagy, networks between lncRNA and mRNA might be crucial for key gene expression, which might help to understand the basic pathophysiological conditions and the development of autophagy-associated DILI.



