




Non-alcoholic fatty liver disease (NAFLD) is the most common liver disorder in industrialized countries and is associated with increased risk of cardiovascular, hepatic and metabolic diseases. Molecular mechanisms on the root of the disrupted lipid homeostasis in NAFLD and potential therapeutic strategies can benefit of in vivo and in vitro experimental models of fatty liver. Here, we describe the high fat diet (HFD)-fed rat in vivo model, and two in vitro models, the primary cultured rat fatty hepatocytes or the FaO rat hepatoma fatty cells, mimicking human NAFLD. Liver steatosis was invariably associated with increased number/size of lipid droplets (LDs) and modulation of expression of genes coding for key genes of lipid metabolism such as peroxisome proliferator-activated receptors (Ppars) and perilipins (Plins). In these models, we tested the anti-steatotic effects of 3,5-L-diiodothyronine (T2), a metabolite of thyroid hormones. T2 markedly reduced triglyceride content and LD size acting on mRNA expression of both Ppars and Plins. T2 also stimulated mitochondrial oxidative metabolism of fatty acids. We conclude that in vivo and especially in vitro models of NAFLD are valuable tools to screen a large number of compounds counteracting the deleterious effect of liver steatosis. Because of the high and negative impact of liver steatosis on human health, ongoing experimental studies from our group are unravelling the ultimate translational value of such cellular models of NAFLD.
Liver steatosis is the consequence of ectopic accumulation of excessive triacylglycerol (TAG) content (> 5% of liver volume or weight)1 in the liver, histologically seen when intracellular TAGs in more than 5% of the hepato-cytes.2 It is the result of an unbalance in lipid hepatic metabolic pathways following four possible mechanisms:
- •
Increased delivery and/or uptake of fatty acids (FAs) or carbohydrates due to excess dietary intake or release from adipose tissue.
- •
Increased de novo lipogenesis (DNL).
- •
Decreased very low-density lipoprotein (VLDL) synthesis and TAG export.
- •
Decreased FA catabolism due to impaired oxidation (Figure 1).
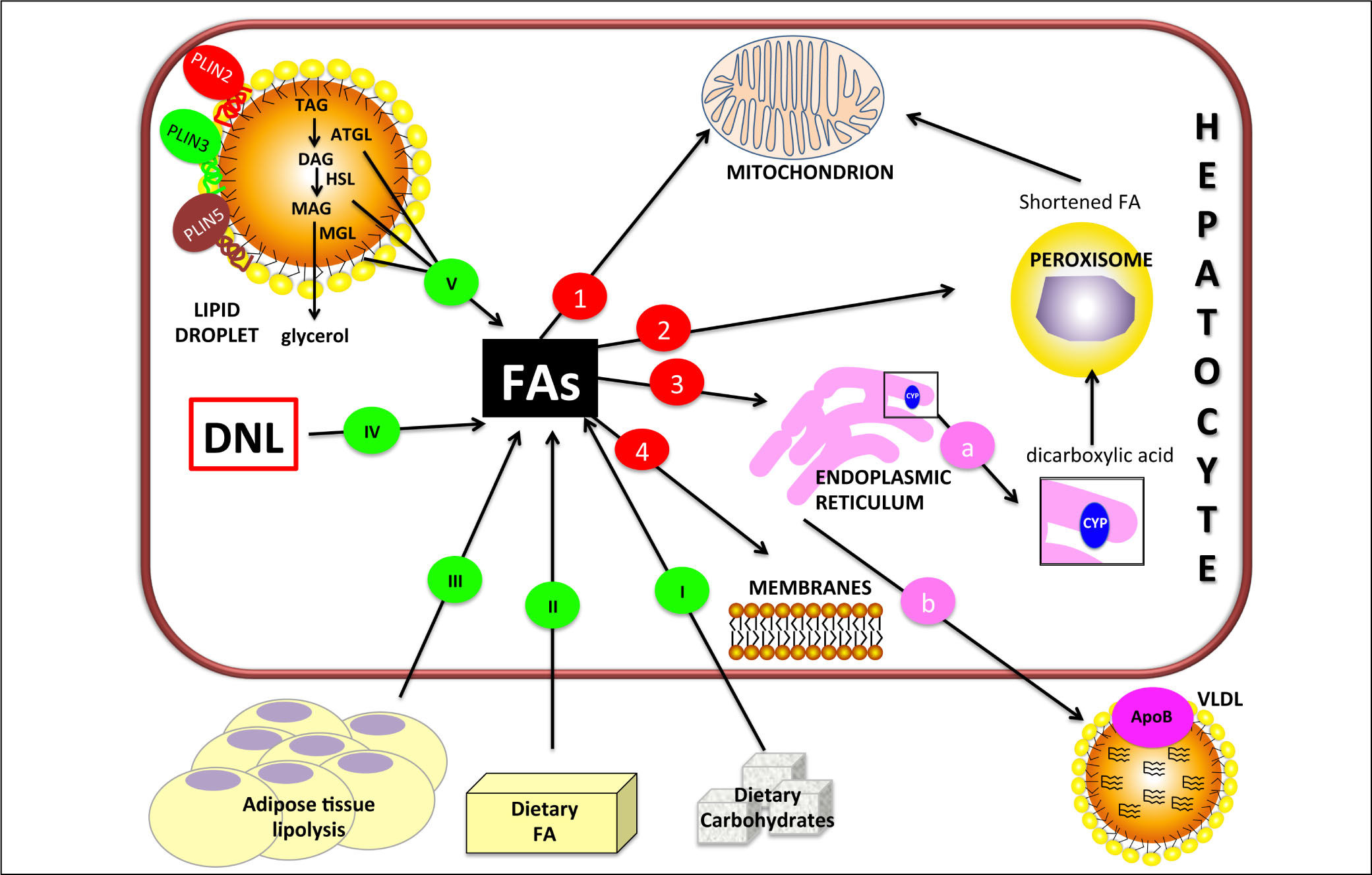
 can derive from external sources such as dietary carbohy drates (I) and/or lipids (II) and also from hydrolysis of fat in adipose tissue (III). Internal sources are de novo lipogenesis (DNL; IV) and release from storage organelles (V). In the cell, FAs are stored in lipid droplet (LD) core mainly in the form of triglycerides (TAGs). LD surface is decorated with an array of proteins including members of the perilipin (PLIN) family, of which PLIN2, PLIN3 and PLIN5 are expressed in normal liver. Lipases are recruited on LD surface and this triggers TAG hydrolysis that occurs sequentially in 3 steps: TAGs are hydrolyzed to form diacylglycerol (DAG) and then monoacylglycerol (MAG), with the release of a FA at each step. These two reactions are catalyzed by adipose triglyceride lipase (ATGL) and hormone sensitive lipase (HSL) respectively. Finally, MAG is hydrolyzed by monoglyceride lipase (MGL) to release the last FA and glycerol. Glycerol can enter glycolytic or gluconeogenic pathways. FAs released from TAG hydrolysis can be channelled to different organelles: 1. Mitochondria: short-chain (< C8), medium-chain (C8-C12), and long-chain (C12-C20) FAs, once transported by the carnitine system, undergo β-oxidation. 2. Peroxisomes: very-long-chain (> C20) FAs undergo β-oxidation; shortened FAs released from peroxisomes are subsequently oxidized in mitochondria. 3. Endoplasmic reticulum: a. in fatty liver, ω-oxidation, a minor pathway of FA metabolism, can be activated when the mitochondrial oxidation system is inadequate to metabolize excess FAs. ω-oxidation produces long-chain dicarboxylic acids by CYP4A subfamily catalysis. Long-chain dicarboxylic acids released from endoplasmic reticulum are subjected to further oxidation steps in peroxisomes. b. FAs can be again esterified and the resulting TAGs are packaged into ApoB containing lipoproteins (VLDL) and consequently secreted from the cell. 4. Membranes: FAs can form phospholipids for membrane synthesis and repair.") Figure 1.
Figure 1.Hepatic lipid metabolism/homeostasis. Free fatty acids (FAs) can derive from external sources such as dietary carbohy drates (I) and/or lipids (II) and also from hydrolysis of fat in adipose tissue (III). Internal sources are de novo lipogenesis (DNL; IV) and release from storage organelles (V). In the cell, FAs are stored in lipid droplet (LD) core mainly in the form of triglycerides (TAGs). LD surface is decorated with an array of proteins including members of the perilipin (PLIN) family, of which PLIN2, PLIN3 and PLIN5 are expressed in normal liver. Lipases are recruited on LD surface and this triggers TAG hydrolysis that occurs sequentially in 3 steps: TAGs are hydrolyzed to form diacylglycerol (DAG) and then monoacylglycerol (MAG), with the release of a FA at each step. These two reactions are catalyzed by adipose triglyceride lipase (ATGL) and hormone sensitive lipase (HSL) respectively. Finally, MAG is hydrolyzed by monoglyceride lipase (MGL) to release the last FA and glycerol. Glycerol can enter glycolytic or gluconeogenic pathways. FAs released from TAG hydrolysis can be channelled to different organelles: 1. Mitochondria: short-chain (< C8), medium-chain (C8-C12), and long-chain (C12-C20) FAs, once transported by the carnitine system, undergo β-oxidation. 2. Peroxisomes: very-long-chain (> C20) FAs undergo β-oxidation; shortened FAs released from peroxisomes are subsequently oxidized in mitochondria. 3. Endoplasmic reticulum: a. in fatty liver, ω-oxidation, a minor pathway of FA metabolism, can be activated when the mitochondrial oxidation system is inadequate to metabolize excess FAs. ω-oxidation produces long-chain dicarboxylic acids by CYP4A subfamily catalysis. Long-chain dicarboxylic acids released from endoplasmic reticulum are subjected to further oxidation steps in peroxisomes. b. FAs can be again esterified and the resulting TAGs are packaged into ApoB containing lipoproteins (VLDL) and consequently secreted from the cell. 4. Membranes: FAs can form phospholipids for membrane synthesis and repair.
(0.29MB).
 can derive from external sources such as dietary carbohy drates (I) and/or lipids (II) and also from hydrolysis of fat in adipose tissue (III). Internal sources are de novo lipogenesis (DNL; IV) and release from storage organelles (V). In the cell, FAs are stored in lipid droplet (LD) core mainly in the form of triglycerides (TAGs). LD surface is decorated with an array of proteins including members of the perilipin (PLIN) family, of which PLIN2, PLIN3 and PLIN5 are expressed in normal liver. Lipases are recruited on LD surface and this triggers TAG hydrolysis that occurs sequentially in 3 steps: TAGs are hydrolyzed to form diacylglycerol (DAG) and then monoacylglycerol (MAG), with the release of a FA at each step. These two reactions are catalyzed by adipose triglyceride lipase (ATGL) and hormone sensitive lipase (HSL) respectively. Finally, MAG is hydrolyzed by monoglyceride lipase (MGL) to release the last FA and glycerol. Glycerol can enter glycolytic or gluconeogenic pathways. FAs released from TAG hydrolysis can be channelled to different organelles: 1. Mitochondria: short-chain (< C8), medium-chain (C8-C12), and long-chain (C12-C20) FAs, once transported by the carnitine system, undergo β-oxidation. 2. Peroxisomes: very-long-chain (> C20) FAs undergo β-oxidation; shortened FAs released from peroxisomes are subsequently oxidized in mitochondria. 3. Endoplasmic reticulum: a. in fatty liver, ω-oxidation, a minor pathway of FA metabolism, can be activated when the mitochondrial oxidation system is inadequate to metabolize excess FAs. ω-oxidation produces long-chain dicarboxylic acids by CYP4A subfamily catalysis. Long-chain dicarboxylic acids released from endoplasmic reticulum are subjected to further oxidation steps in peroxisomes. b. FAs can be again esterified and the resulting TAGs are packaged into ApoB containing lipoproteins (VLDL) and consequently secreted from the cell. 4. Membranes: FAs can form phospholipids for membrane synthesis and repair.")
Non-alcoholic fatty liver disease (NAFLD) is considered the hepatic phenotype of the metabolic syndrome (MS), associated with insulin resistance or established type 2 diabetes, increased visceral adiposity, overweight/ obesity, dyslipidemia and blood hypertension.3 NAFLD is associated with increased risk of cardiovascular, hepatic and metabolic diseases and is characterized by excessive TAG accumulation in the absence of other causes of chronic liver disease, such as hepatotoxic xenobiotics (alcohol/drugs) and viral infections. NAFLD is recognized as the leading cause of chronic liver disease in adults and children4 and has an estimated prevalence of 20-40% in Western countries.5 The current Western diet, high in saturated fats and fructose, plays a significant role in the pathogenesis of NAFLD.6 NAFLD encompasses a wide spectrum of diseases ranging from simple steatosis to non-alcoholic steatohepatitis (NASH) which can progress to more severe pathologies such as cirrhosis.7
At present, specific targeted pharmacological therapies are still lacking and the mainstay of therapy remains weight loss through lifestyle changes, including dietary modification and regular physical activity.5,8 The understanding of the molecular pathways related to NAFLD is useful to avoid its harmful progression and represents the basis for finding new therapeutic strategies.9 Experimental models of NAFLD represent effectual tools for research in this field, and our group is actively involved in this respect.
Here, we provide a brief overview on the role of per-oxisome proliferator-activated receptors (PPARs) and li-pid droplet (LD)-associated perilipins (PLINs) in lipid homeostasis. We summarise and compare the features of the in vivo and in vitro models of NAFLD widely employed by our group. Both models were employed to test the therapeutic potential of a thyroid hormone derivative, 3,5-L-diiodothyronine, acting as a modulator of hepatic lipid metabolism. In perspective, similar models represent valuable tools to screen a large number of compounds counteracting the deleterious effect of liver steatosis.
Hepatic Lipid Homeostasis in Health and DiseaseThe key role of the liver in lipid homeostasis is maintained throughout evolution. The major lipid metabolic pathways in hepatocytes are summarised in Figure 1.8
In mammals, the adipose tissue stores excess lipids in the form of TAGs in cytosolic LDs. LDs are present also in hepatocytes, heart and skeletal myocytes, adrenocortical cells, enterocytes, and macrophages.10 LDs share their structural features with lipoproteins: both contain a neutral lipid core encased in a polar lipid monolayer, decorated by specific proteins. In the past, LDs were considered as mere fat depots, but they are now recognized as dynamic or-ganelles at the hub of lipid and energy metabolism.11 The dynamic features of LDs are confirmed by changes in the expression of their proteome that reflects the metabolic status of the cell.12 The most documented group of LD-asso-ciated proteins is the PLIN family,13 which comprises: perilipin (PLIN1), adipophilin/ADRP (adipose differentiation related protein; PLIN2), TIP47 (tail-interacting protein of 47 kDa; PLIN3), S3-12 (PLIN4) and OXPAT (oxidative tissue-enriched PAT protein; PLIN5).14 The main function of PLINs is to regulate LD metabolism by selectively recruiting lipases and other proteins to LD surface. Their sequence features are depicted in figure 2A: a common feature of all PLINs is the propensity to coat LDs, whereas sequence differences confer specificity of function, transcriptional regulation and tissue expression.15 In the liver, PLINs are expressed with differences among humans and rodents. For example, PLIN1 is absent in normal and steatotic livers of mice,16 but it was found in hepatocytes isolated from humans with NAFLD. PLIN2 is constitu-tively expressed in human and murine liver. Expression of PLIN3 and PLIN5 has been widely documented in human and mouse liver, and, more recently, in rat hepatocytes.17 Upregulation of PLIN2 has been reported in the liver of rodents and humans with NAFLD.18,19 Actually, PLIN2 promotes TAG accumulation,20 inhibits FA oxidation,21 and impairs glucose tolerance.19,22Plin2 knock-out mice are protected from hepatic steatosis when fed a HFD.23 PLIN3 is upregulated in fatty livers of HFD-fed mice and Plin3 anti-sense oligonucleotide reduces hepatic TAGs and improves insulin sensitivity.24 Expression of PLIN5 is increased in fatty livers of dystrophic mice,25 and Plin5 knock-out mice are protected from hepatic steatosis due to increased lipolysis and fat oxidation.26
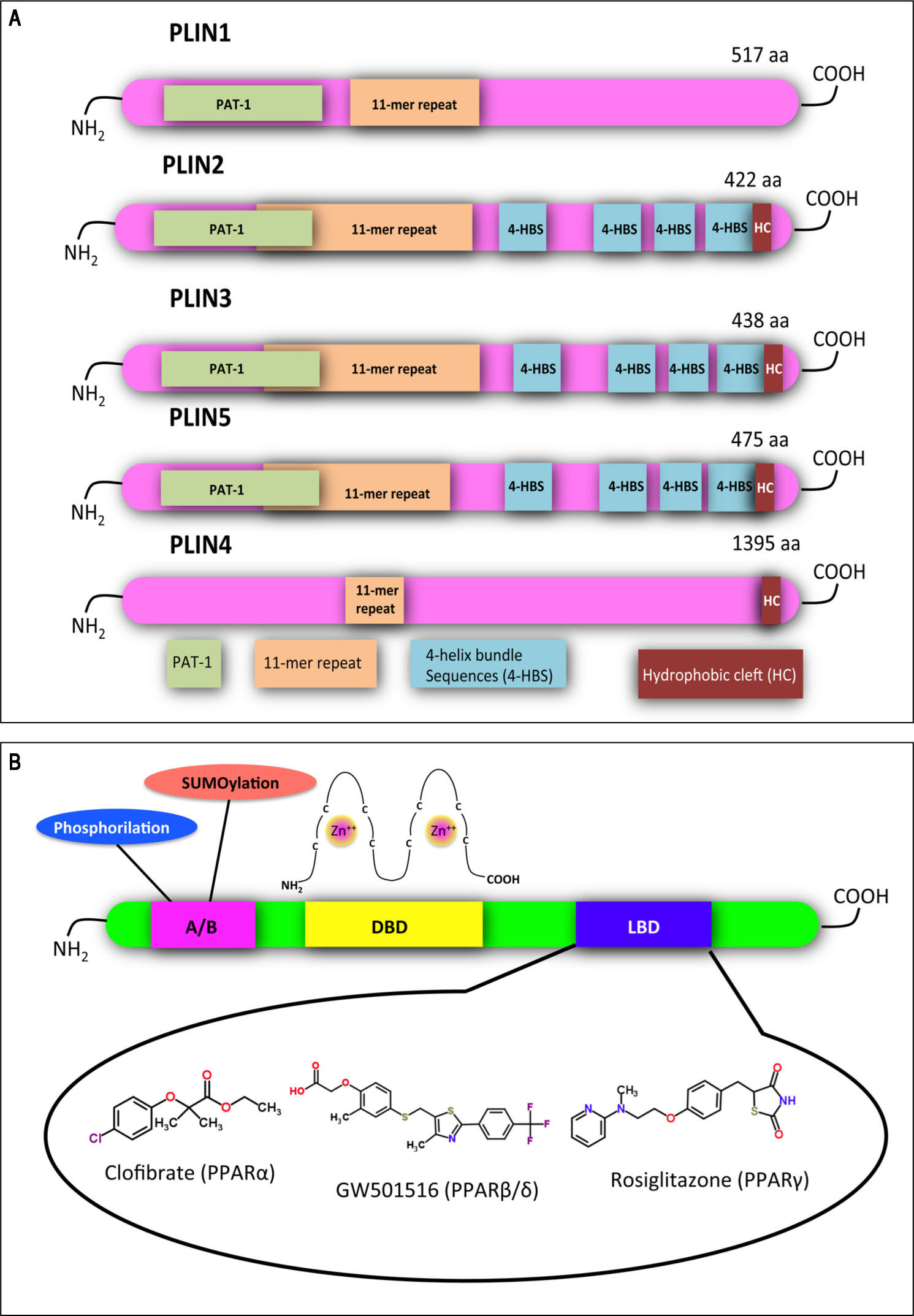
. Despite the absence of a clear PAT domain, PLIN4 retains sequence similarity to the other family members in other parts of the protein, including the 11-mer repeats (orange). X-ray crystallography has revealed a four-helix bundle sequence in PLIN3 (4-HBS, blue), which has been predicted also in PLIN2 and PLIN5. This domain is proposed to target the proteins to LDs. A hydrophobic cleft (HC, brown) is located at the C-termini of PLIN2, 3, 4 and 5. Adapted from references14, 84. B. Functional domains found in PPARs and other nuclear receptors: N-terminal A/B domain (pink); DNA-bind-ing domain (DBD, yellow); ligand binding domain (LBD, blue). The DBD contains two zinc finger motifs, which bind to specific sequences of DNA known as peroxisome proliferator response element (PPRE) when the receptor is activated. Selective agonists for the 3 PPAR isoforms are indicated.")
Structural features of PLINs and PPARs. A. A schematic diagram of the structural features and the amino acid sequence similarities of PLINs. A ~100 amino acid region of high sequence similarity near their N-termini is defined as the PAT domain (PAT-1, green). Despite the absence of a clear PAT domain, PLIN4 retains sequence similarity to the other family members in other parts of the protein, including the 11-mer repeats (orange). X-ray crystallography has revealed a four-helix bundle sequence in PLIN3 (4-HBS, blue), which has been predicted also in PLIN2 and PLIN5. This domain is proposed to target the proteins to LDs. A hydrophobic cleft (HC, brown) is located at the C-termini of PLIN2, 3, 4 and 5. Adapted from references14, 84. B. Functional domains found in PPARs and other nuclear receptors: N-terminal A/B domain (pink); DNA-bind-ing domain (DBD, yellow); ligand binding domain (LBD, blue). The DBD contains two zinc finger motifs, which bind to specific sequences of DNA known as peroxisome proliferator response element (PPRE) when the receptor is activated. Selective agonists for the 3 PPAR isoforms are indicated.
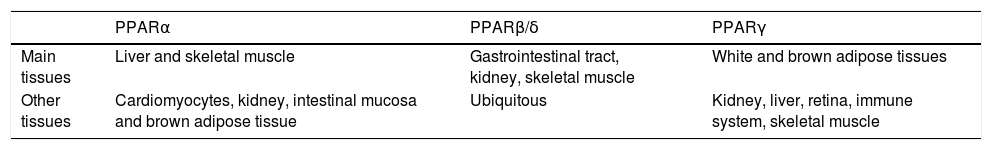
In the hepatocyte, genes coding for LD-associated Plins are under control of several transcription factors, among which a pivotal role is played by PPARs, that are ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily.27 To date, three PPAR iso-forms have been identified: PPARcc, PPAR(3/8 and PPARy. They share common structural features, which include an amino-terminal modulatory domain, a DNA binding domain, a carboxyl-terminal ligand-binding domain, and the ability to form heterodimers with the retinoid X receptor. Activated heterodimers bind to peroxisome proliferator response element (PPRE) in the promoter region of target genes 28 (Figure 2B). Many natural and synthetic compounds have been identified as PPAR ligands, and this underlines the important role of PPARs as therapeutic targets.29 PPAR isoforms exhibit specific but partially overlapping tissue expression patterns (Table 1). In the liver, PPARcc is the master regulator of both (3- and co-oxidation and PPAR(3/8 is also required for hepatic FA oxidation.30 Conversely, PPARy expression is induced when energy storage is required.8
Tissue expression of peroxisome proliferator-activated receptor (PPAR) isoforms. Modified from reference 85.
| PPARα | PPARβ/δ | PPARγ | |
|---|---|---|---|
| Main tissues | Liver and skeletal muscle | Gastrointestinal tract, kidney, skeletal muscle | White and brown adipose tissues |
| Other tissues | Cardiomyocytes, kidney, intestinal mucosa and brown adipose tissue | Ubiquitous | Kidney, liver, retina, immune system, skeletal muscle |
PPARcc expression is upregulated in the liver of high-fat diet (HFD)-fed rodents and in obese Wistar rats,31-33 as well as in human NAFLD biopsies,34 even if in some models it is reported to be decreased, possibly depending on different diet compositions and/or durations.35-36 PPARy mediates the development of liver steatosis by mechanisms involving activation of lipogenic genes, as demonstrated both in animal models of obesity,37 and in obese patients with NAFLD.38 Treatment of ob/ob mice with the PPARy agonist rosiglitazone increased oxidative stress and liver steatosis.39 Regarding PPAR(3/8, its contribution to NAFLD is still unclear. It has been reported that treatment with the PPAR(3/8 agonist GW501516 ameliorates hepatic steatosis and inflammation in the methionine-choline deficient (MCD) diet induced-mouse model of NASH40.
As aforementioned, genes coding for LD associated proteins are PPAR targets. Particularly, PPARy appears to regulate the expression of Plin1, Plin2, Plin4 and Plin5 in adipose tissue, and Plin2 and Plin5 in liver.41-43 Moreover, the pharmacological PPARcc agonist Wy-14643 is able to stimulate the expression of Plin5 in liver, heart, and skeletal muscle.42,44 PPRE responsive to PPARcc and PPAR(3/8 were found upstream of Plin2 coding gene in both rat and human hepatocyte-derived cell lines.45
In Vivo Models of Nafld: The hfd-Fed RatInvestigating the pathophysiology and possible treatments of NAFLD in humans is limited by the long time required for the occurrence of steatosis and the progression to NASH, and also by ethical concerns regarding tissue collection and testing of drugs. Reliable and simple animal models resembling the most important features of NAFLD are needed. Even though some studies have proposed non-rodent mammalian models, the most used are still those employing mouse and rat.46,47
In vivo NAFLD models are represented by two main categories: genetic and dietary. The most frequent genetic models are the spontaneously diabetic rodents deficient in leptin or leptin receptors, which include ob/ob mouse, db/db mouse and Zucker fatty rat.48 Rodent dietary models encompass several treatments that differ in composition and duration, such as methionine and choline deficient (MCD) diet, high-fructose and/or sucrose diet, cafeteria diet, high cholesterol diet and HFD.49 Diversities in dietary models also regard strain, sex and age of animals, the amount of energy derived from fats, the origin of fats, the ratio of saturated (SFA) to monounsaturated (MUFA) and polyunsaturated (PUFA) fatty acids, and the proportion between ω-3 and ω-6 PUFA. Such differences are reflected in outcome variations ranging from mild hepatic steato-sis to severe NASH.47
In our studies, we utilized male Wistar rats, which were administered for one month with a HFD (for a detailed description of the HFD please refer to figure 3A legend).50,51 At the end of treatment, hepatic lipid accumulation was demonstrated by staining of liver sections with fat-soluble Oil red-O (ORO), that revealed the presence of numerous LDs indicating the microvesicular steatosis typical of NAFLD (Figure 4A, left panel).
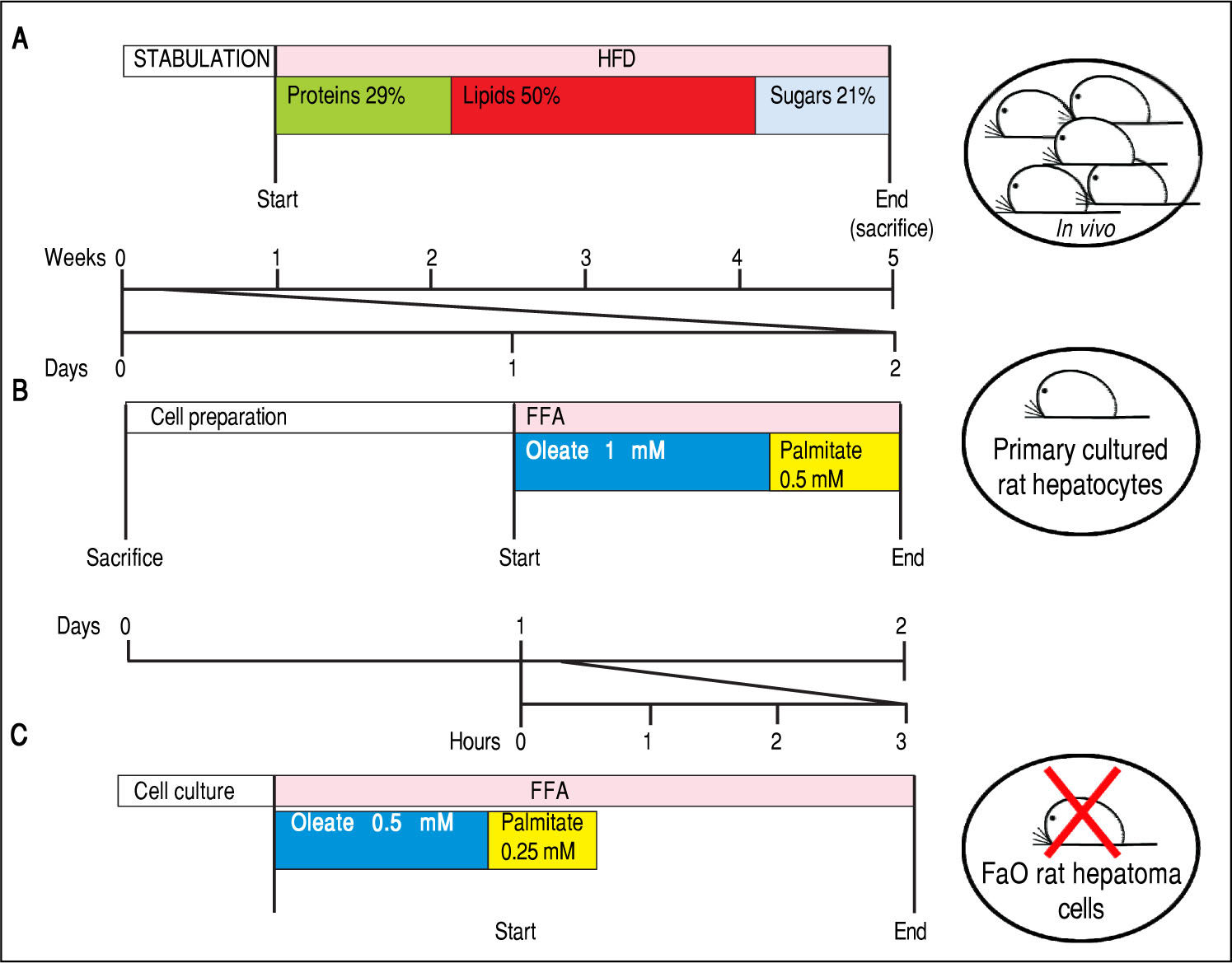
. Male Wistar rats (aged 8 weeks) were housed in individual cages in a temperature-controlled room at 28 oC with a 12:12-h light:dark cycle. After one week of acclimation, the animals were administered the HFD where most of the total metabolizable percentage of energy derived from fats with respect to sugars (21% carbohydrates, 29% proteins, 50% lipids (19.85 kJ gross energy/g).50,51 In the standard control diet, total metabolizable percentages of energy were: 60.4% carbo-hydrates, 29% proteins, 10.6% lipids (15.88 kJ gross energy/g). At least 5 animals must be included in each experimental group (Control and HFD-fed). At the end of the treatments, all rats were sacrificed. The total time required to obtain hepatocellular steatosis was 5 weeks. B. In vitro model of NAFLD using rat hepatocyte primary cultures. Hepatocytes were isolated from only one animal, plated and allow to attach and restore. The day after, cells were treated with oleate:palmitate (FA) mixture 2:1 (1.5 mM) for 24 h. The total time required to obtain hepatocellular steatosis was 48 h. C. In vitro model of NAFLD using FaO rat hepatoma cell line. Cells were routinely passaged and when at 70-80% confluence they were incubated with oleate:palmitate (FA) mixture 2:1; (0.75 mM) for 3 h. No rats were sacrificed and the total time required to obtain hepatocellular steatosis was 3 h.")
Experimental protocols for in vivo and in vitro models of NAFLD. In this figure we compare the number of animals needed and the duration of the treatments used in vivo and in vitro to obtain our 3 different models of NAFLD: a dramatic decrease in animal use and a high speeding up of the experimental procedures can be appreciated. A. In vivo model of NAFLD (HFD). Male Wistar rats (aged 8 weeks) were housed in individual cages in a temperature-controlled room at 28 oC with a 12:12-h light:dark cycle. After one week of acclimation, the animals were administered the HFD where most of the total metabolizable percentage of energy derived from fats with respect to sugars (21% carbohydrates, 29% proteins, 50% lipids (19.85 kJ gross energy/g).50,51 In the standard control diet, total metabolizable percentages of energy were: 60.4% carbo-hydrates, 29% proteins, 10.6% lipids (15.88 kJ gross energy/g). At least 5 animals must be included in each experimental group (Control and HFD-fed). At the end of the treatments, all rats were sacrificed. The total time required to obtain hepatocellular steatosis was 5 weeks. B. In vitro model of NAFLD using rat hepatocyte primary cultures. Hepatocytes were isolated from only one animal, plated and allow to attach and restore. The day after, cells were treated with oleate:palmitate (FA) mixture 2:1 (1.5 mM) for 24 h. The total time required to obtain hepatocellular steatosis was 48 h. C. In vitro model of NAFLD using FaO rat hepatoma cell line. Cells were routinely passaged and when at 70-80% confluence they were incubated with oleate:palmitate (FA) mixture 2:1; (0.75 mM) for 3 h. No rats were sacrificed and the total time required to obtain hepatocellular steatosis was 3 h.
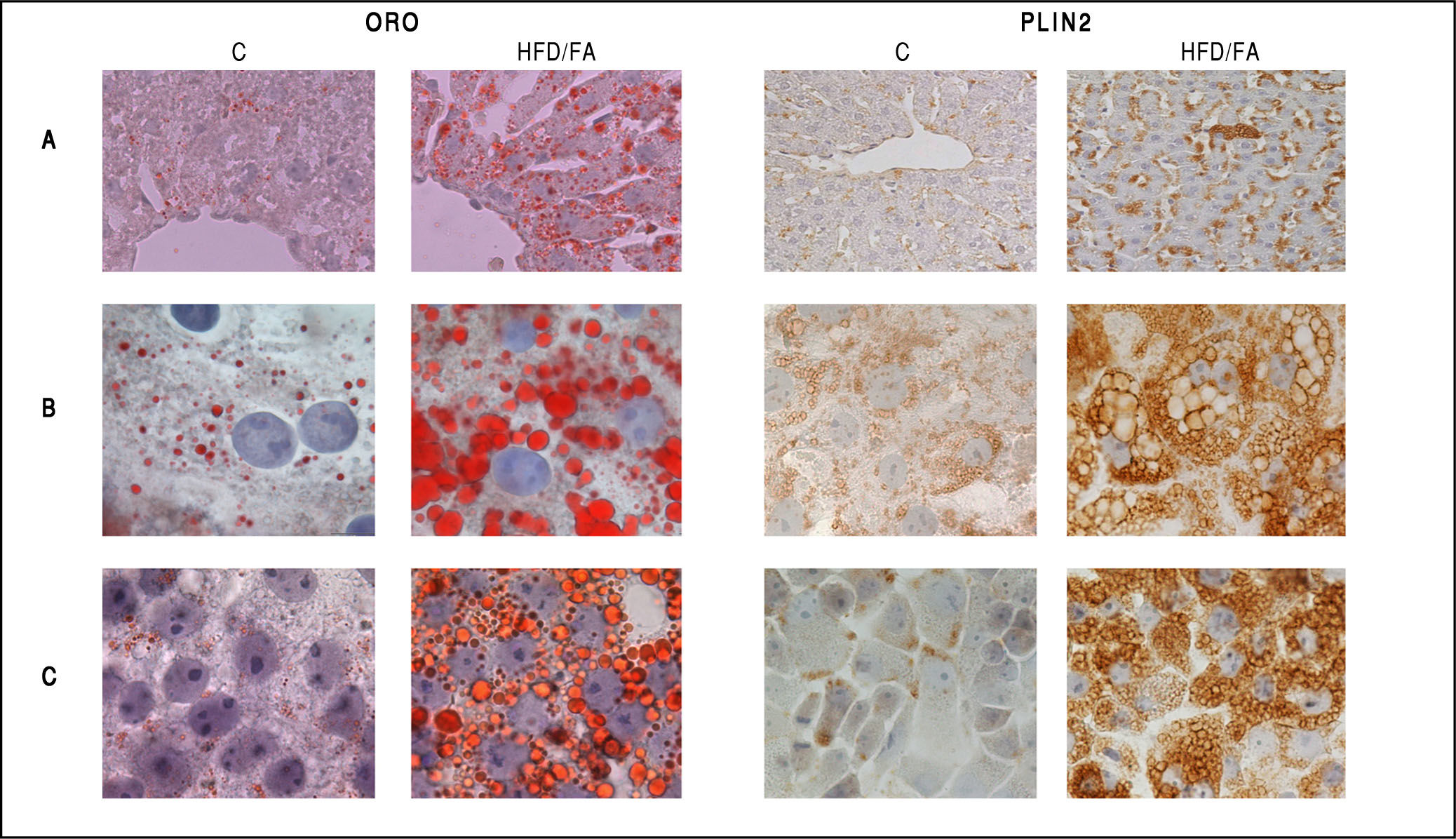
 and PLIN2 (right panel) staining in control (C) and steatotic hepatocytes (HFD/FA) in liver tissue sections (A), primary cultured hepatocytes (B) and FaO rat hepatoma cells (C). Lipid accumulation as well as increase in PLIN2-positive vesicles is evident for all NAFLD models. Immunostaining was performed with anti-PLIN2 polyclonal antibody (clone GP40-mN1, Fitzgerald Industries International, Concord, MA, USA) (magnification 100X).")
Neutral lipid accumulation and PLIN2 immunostaining in in vivo and in vitro models of NAFLD. Oil red-O (ORO, left panel) and PLIN2 (right panel) staining in control (C) and steatotic hepatocytes (HFD/FA) in liver tissue sections (A), primary cultured hepatocytes (B) and FaO rat hepatoma cells (C). Lipid accumulation as well as increase in PLIN2-positive vesicles is evident for all NAFLD models. Immunostaining was performed with anti-PLIN2 polyclonal antibody (clone GP40-mN1, Fitzgerald Industries International, Concord, MA, USA) (magnification 100X).
Several in vitro models of NAFLD have been developed, mainly consisting of primary hepatocyte cultures52,53 or hepatocyte cell lines54 treated with monounsaturated and/or saturated FAs (MUFAs and SFAs, respectively). These models usually employ palmitate (C16:0) or oleate (C18:1), or a mixture of the two,55 which are common long chain FAs in the Western diet56 and the most abundant FAs in liver of both normal subjects and patients with NAFLD.57 Adding oleate to palmitate-treated cells can fully prevent palmitate lipotoxicity together with enhanced TAG synthesis.58 In this light, we used oleate:palmitate mixture in a 2:1 molar ratio to developed in vitro models of NAFLD in rat hepatic cells. For better resembling in vivo conditions, we chose FA concentrations comparable to the plasmatic levels of patients with MS.59
Rat hepatocyte primary culturesRat hepatocyte primary cultures have been extensively used in basic research on liver function.60 To isolate intact hepatocytes,61 we basically applied the method of Seglen,62 followed by maintenance of cells in appropriate culture conditions.63 We cultured rat hepatocytes in serum-free DMEM containing 0.25% bovine serum albumin (BSA). After 24 h of adaptation to the in vitro environment, hepato-cytes were exposed to a mixture of oleate/palmitate (2:1 molar ratio, total concentration 1.5 mM) for 24 h17,64 (Figure 3B and 4B, left panel).
Primary hepatocytes do not proliferate in vitro and can be used only within the first week of culture, because over time they tend to lose the differentiated adult liver phenotype. Cells need to be freshly isolated for each experiment, but a pure hepatocyte population (100-400 millions) with high viability is routinely obtained from each animal. Hepatocellular steatosis in primary cultures is achieved within only 1 day of treatment with FA mixture; in contrast, 4 weeks of HFD administration to rats are necessary to induce NAFLD in vivo (Figure 3B and 4B left panel).
Rat hepatoma cell lineThe FaO rat hepatoma cells express a broad array of liver-specific mRNAs and maintain the ability to assemble and secrete VLDL65,66 as well as to respond to stimuli that activate PPARs.67 In our experiments, FaO cells at 70-80% confluence were incubated in starvation medium (F12 Coon's modified medium supplemented with 0.25% BSA) with a mixture of oleate/palmitate (2:1 molar ratio, total concentration 0.75 mM). It is noteworthy that basal lipid content of FaO cells is much lower than that of primary hepatocytes, thus, FA treatment resulted in a more rapid and pronounced TAG accumulation with respect to primary cultured hepatocytes reaching a plateau at 3-6 h68 (Figure 3C). Moreover, the effect was induced using a lower concentration of FA mixture (0.75 mM vs. 1.5 mM). In these conditions, a marked increase in number and size of cytosolic LDs was observed by ORO staining (Figure 4C, left panel). These observations candidate lipid-loaded FaO cells as a reliable and convenient in vitro model of NAFLD.
Expression of Lipid Metabolism Genes and Secretion of Lipids in Models of NafldThe expression of Plins and Ppars, as well as TAG secretion, were altered in both in vitro and in vivo models of NAFLD. Our findings are summarized in table 2.51,64,68-71
Expression of Plins and Ppars and TAG secretion in in vivo and in vitro models of NAFLD. Changes in mRNA expression levels of perilipin (Plins) proteins (Plin2; Plin3; Plin5) and isoforms of peroxisome proliferator activated receptors (Ppary; Pparcc; Ppar p/8), in three different in vivo and in vitro models of NAFLD. Changes in triglyceride (TAG) secretion are also shown. Significant changes are reported as percentage with respect to controls and “-” indicates no changes.
Changes in Plin expression resulted fully consistent in all models. Plin5 and Plin2 mRNA levels were always significantly increased compared to controls. In particular, im-munohistochemical staining of PLIN2 revealed little or absent PLIN2 immunoreactivity in controls cells of all models. Lipid accumulation in hepatocytes was accompanied by a dramatic increase in size and number of PLIN2-positive vesicles both in vivo and in vitro (Figures 4A-4C; right panel). No changes in the expression of Plin3 were observed in either steatotic liver or fatty hepatocytes. However, an increased expression of Plin3 was observed in primary hepatocytes at shorter (12 h) times of FA exposure.17 Therefore, induction of Plin3 may represent a transient and short-time response of the hepatocyte to excess FAs.
Also expression of the different Ppar isoforms was modified in all models of NAFLD (Table 2). High Pparγ expression is one of the main features of the steatotic liver.37,38 Accordingly, increased Ppary expression was a common trait observed in all experimental models. Ppara is mainly involved in regulation of lipid oxidation. HFD provides excess of long-chain FAs entering the hepato-cytes with consequent activation of Ppara and upregula-tion of those PPARcc-target genes involved in FA catabolism.72 We observed that Ppara expression was increased in the liver of HFD rats, whereas no changes were recorded in vitro. These differences may be ascribed to the experimental conditions utilized to induce lipid accumulation in vivo and in vitro. Moreover, in NAFLD animal models, some authors documented a decrease in Ppara expression related to an impaired mitochondrial FA oxidation and to a decrease in carnitine-palmitoyl-transferase (Cpt1) expression.27 Accordingly, in HFD-fed rats, the increased Ppara expression was associated with an increase in Cpt1 enzymatic activity.73 It appears that diet duration can play an important role in determining the dynamic equilibrium of Ppar regulation. De Lange, et al.74 documented increased expressions of Ppara and Ppar/3/8 after 2 weeks of HFD administration, which could be a compensatory mechanism since it was abolished after 4 weeks.
Regarding Ppar/3/8 mRNA levels, they were not affected in HFD rats, but they were upregulated in steatotic primary hepatocytes, and down-regulated in FaO cells. These discrepancies further underline the uncertainties about the role of Ppar/3/8 in NAFLD.
Finally, in the general scenario of the modulation of lipid pathways involved in NAFLD, increasing VLDL secretion can be considered as a compensatory mechanism to overcome lipid overload. Actually, in all experimental models an enhanced lipid secretion was observed: increased levels of TAGs were detected in serum of HFD-rats, as well as in culture media of fatty hepatocytes (Table 2).
Taking into account the differences among the experimental models considered in our studies, the results underline how the main markers of hepatic steatosis are maintained in the in vivo and in vitro models, thus making the simpler in vitro model suitable for rapid study of direct anti-steatotic effects of natural and artificial compounds.
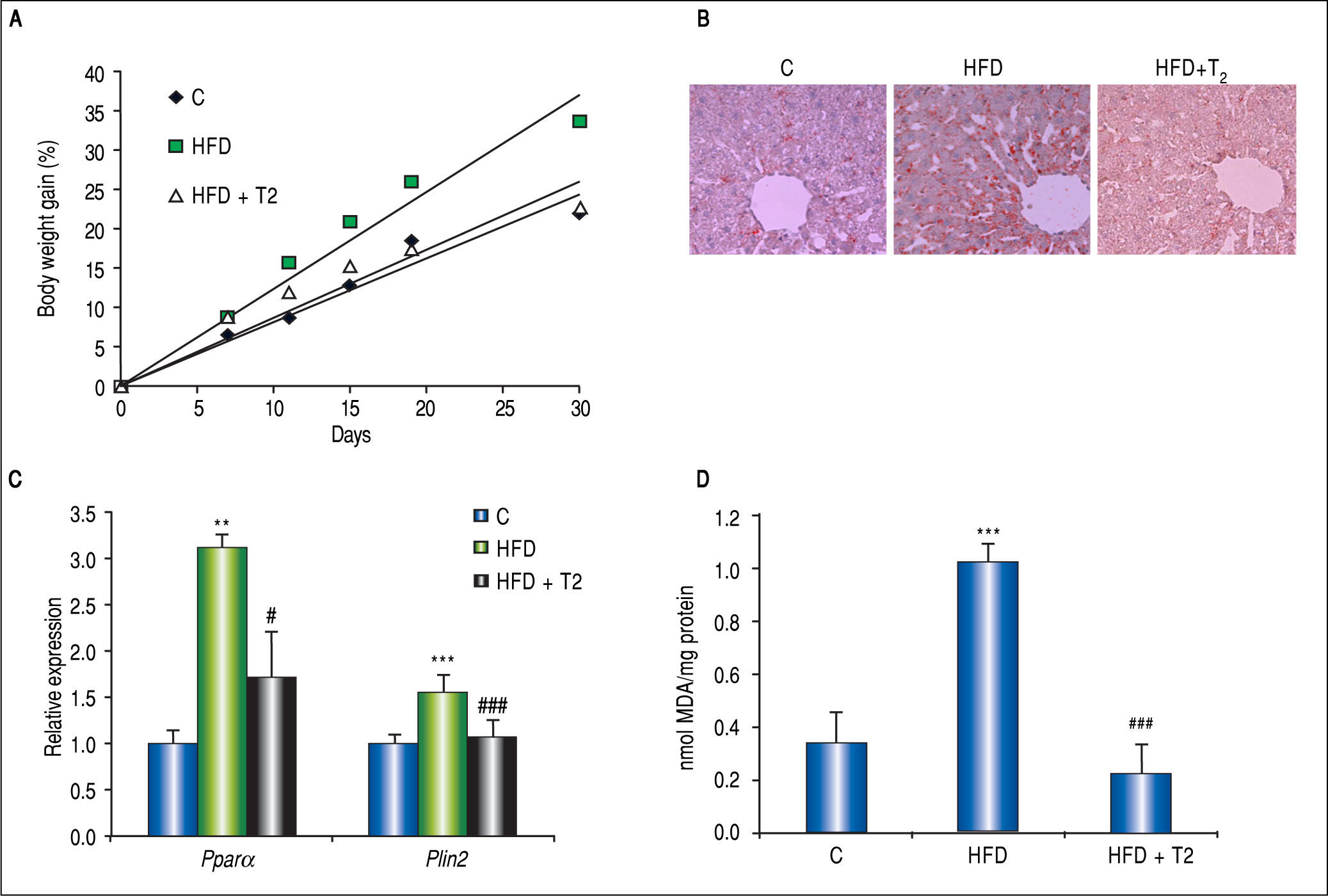
Potential Therapy for Liver Steatosis: The Lipid Lowering Effect of 3,3’,5-L-DiiodothyronineThe liver is a major target for the thyroid hormones (THs) thyroxine (T4) and 3,5-L-diiodothyronine (T3) that play a key role in energy balance and lipid metabolism. High levels of THs stimulate energy expenditure and fat oxidation.75 Studies on humans and animal models indicate that NAFLD is associated with impaired TH signalling.75 The use of THs as potential drugs to treat obesity, hyperlipidemia and MS has been suggested.76 However, due to their collateral dangerous effects, their employment is not recommended. The identification of TH agonists/analogs retaining anti-obesity and hypolipemic efficacies, while being devoid of thyrotox-ic effects, would represent a potential therapeutic advance. Several iodothyronines other than T3 and T4 have been proven to display some thyromimetic activities. Among them, 3,5-L-diiodothyronine (T2) mimics several effects of T3 on energy metabolism without inducing thyrotoxic effects.77 The in vivo HFD model of NAFLD described above has been widely utilized to demonstrate T2 action on hepatic lipid metabo-lism.51,69,73,78,79 Rats receiving T2 along with HFD did not gain weight respect to controls (Figure 5A). In the liver, T2 administration resulted in an almost complete absence of fat accumulation, as revealed by ORO staining (Figure 5B), and HFD-associated lipid peroxidation (Figure 5D); moreover, Pparα and Plin2 mRNA up-regulation in HFD rats was prevented by T2 (Figure 5C). These effects were associated to stimulation of FA oxidation in mitochondria and peroxisomes.72,73
 to rats simultaneously receiving HFD. A. Body weight gain measured throughout the experimental period (30 days) for the 3 experimental groups: C are standard diet-fed rats. Values are expressed as means ± SD of body-weight gain (%). B. ORO staining in liver tissue sections in the 3 experimental groups (magnification 100X). C. Relative mRNA expression of Ppara and Plin2 evaluated by RT-qPCR. Data (mean ± SD, N = 4) are reported as fold induction with respect to controls after normalization for Gapdh mRNA. Significant differences are indicated (C vs. HFD **p < 0.01, ***p < 0.001; HFD vs. HFD + T2 #p < 0.05, ###p < 0.001). D. Liver lipid peroxidation evaluated by TBARs assay. Data (mean ± SD, N = 4) are expressed as nmol MDA/mg protein. Significant differences are indicated (C vs. HFD ***p < 0.001; HFD + T2 vs. FA ###p < 0.001).")
In vivo anti-steatotic effects of T2. T2 was administered daily by i.p. injection (25 fig/100 g bw) to rats simultaneously receiving HFD. A. Body weight gain measured throughout the experimental period (30 days) for the 3 experimental groups: C are standard diet-fed rats. Values are expressed as means ± SD of body-weight gain (%). B. ORO staining in liver tissue sections in the 3 experimental groups (magnification 100X). C. Relative mRNA expression of Ppara and Plin2 evaluated by RT-qPCR. Data (mean ± SD, N = 4) are reported as fold induction with respect to controls after normalization for Gapdh mRNA. Significant differences are indicated (C vs. HFD **p < 0.01, ***p < 0.001; HFD vs. HFD + T2 #p < 0.05, ###p < 0.001). D. Liver lipid peroxidation evaluated by TBARs assay. Data (mean ± SD, N = 4) are expressed as nmol MDA/mg protein. Significant differences are indicated (C vs. HFD ***p < 0.001; HFD + T2 vs. FA ###p < 0.001).
The in vivo studies do not allow to distinguish between the direct anti-steatotic effects of T2 and its indirect effects due to upstream changes in endocrine or metabolic pathways. Moreover, since in vivo T2 was given to rats simultaneously to HFD, the observed effects demonstrated that T2 was able to prevent, rather than to ameliorate, hepatic lipid overload. In order to investigate the direct mechanism of the lipid-lowering effect of T2 on hepatocytes, as well as its capacity to reverse excess fat accumulation, T2 was administered in vitro after lipid-overloading of cells.
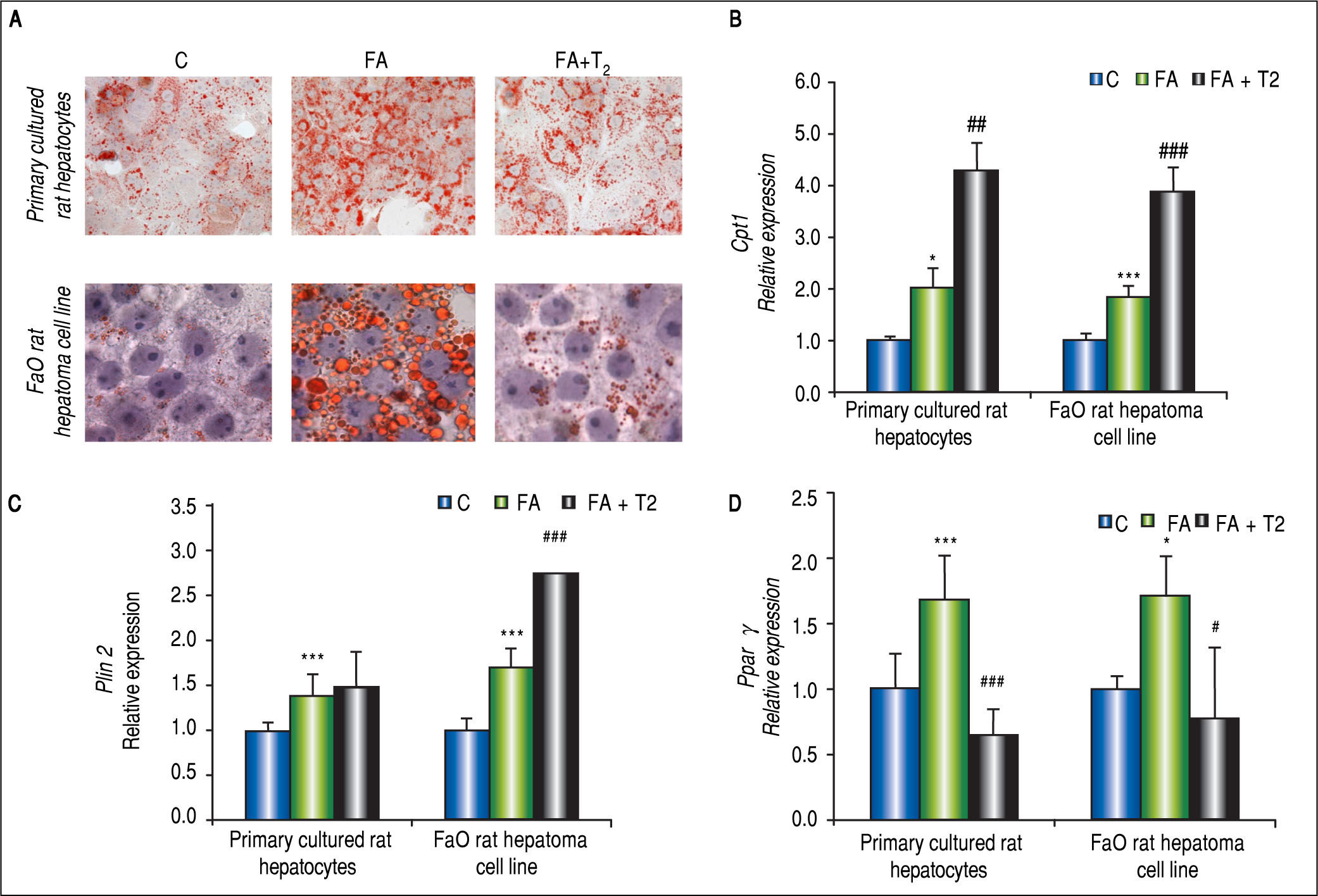
In hepatocyte primary cultures,64 cells were incubated with FA mixture for 24 h; then, the culture medium was replaced and supplemented with T2 at different concentrations (from 10-7 to 10-5 M) for further 24 h. A reduction in number and average size of LDs was observed after treatment with T2 (Figure 6A), suggesting that the iodothyronine led to dispersion/fragmentation of LDs. A decrease in LD size could make the stored TAGs more accessible to enzymes acting on catabolism/ secretion of FAs and this could explain, at least partially, the ability of T2 to decrease the excess fat by acting directly on the hepatic cell. Actually, in steatotic hepa-tocytes T2 was able to reduce lipid accumulation through the recruitment of adipose triglyceride lipase (ATGL) on LD surface, and to down-regulate Plin3.71 The excess FAs produced by the activity of ATGL were mainly channeled into mitochondria for β-oxidation rather than to secretion, as suggested by both up-regula-tion of Cpt1 expression (Figure 6B) and stimulation of cytochrome-c oxidase (COX) activity.71 These data indicate a stimulation of the mitochondrial function, in line with results reported in vivo.73 Regarding Ppars and Plins, T2 was able to reduce the up-regulation of Pparγ induced by FA exposure, whereas Plin2 expression was unaffected (Figures 6C-6D).
 and FA hepatocytes incubated in the absence or in the presence of T2 (FA + T2). Nuclear staining with hematoxylin is also shown (magnification 40X and 100X). B. Relative mRNA expression of Cpt1 evaluated by RT-qPCR. C. Relative mRNA expression of Plin2 evaluated by RT-qPCR. D. Relative mRNA expression of Pparγ evaluated by RT-qPCR. B-D. Data (means ± SD, N = 4) are reported as fold induction with respect to controls after normalization for Gapdh mRNA. Significant differences are indicated (C vs. FA *p < 0.05, ***p < 0.001; FA vs. FA + T2 #p < 0.05, ##p < 0.01, ###p < 0.001).")
In vitro lipid-lowering effects of T2. Primary rat hepatocytes or FaO hepatoma cells were lipid-loaded with FA mixture as described in the text. After 24 h, the culture medium was replaced and supplemented with T2 at 10-5 M and cells were incubated for further 24 h. A. Representative image of ORO-stained control (C) and FA hepatocytes incubated in the absence or in the presence of T2 (FA + T2). Nuclear staining with hematoxylin is also shown (magnification 40X and 100X). B. Relative mRNA expression of Cpt1 evaluated by RT-qPCR. C. Relative mRNA expression of Plin2 evaluated by RT-qPCR. D. Relative mRNA expression of Pparγ evaluated by RT-qPCR. B-D. Data (means ± SD, N = 4) are reported as fold induction with respect to controls after normalization for Gapdh mRNA. Significant differences are indicated (C vs. FA *p < 0.05, ***p < 0.001; FA vs. FA + T2 #p < 0.05, ##p < 0.01, ###p < 0.001).
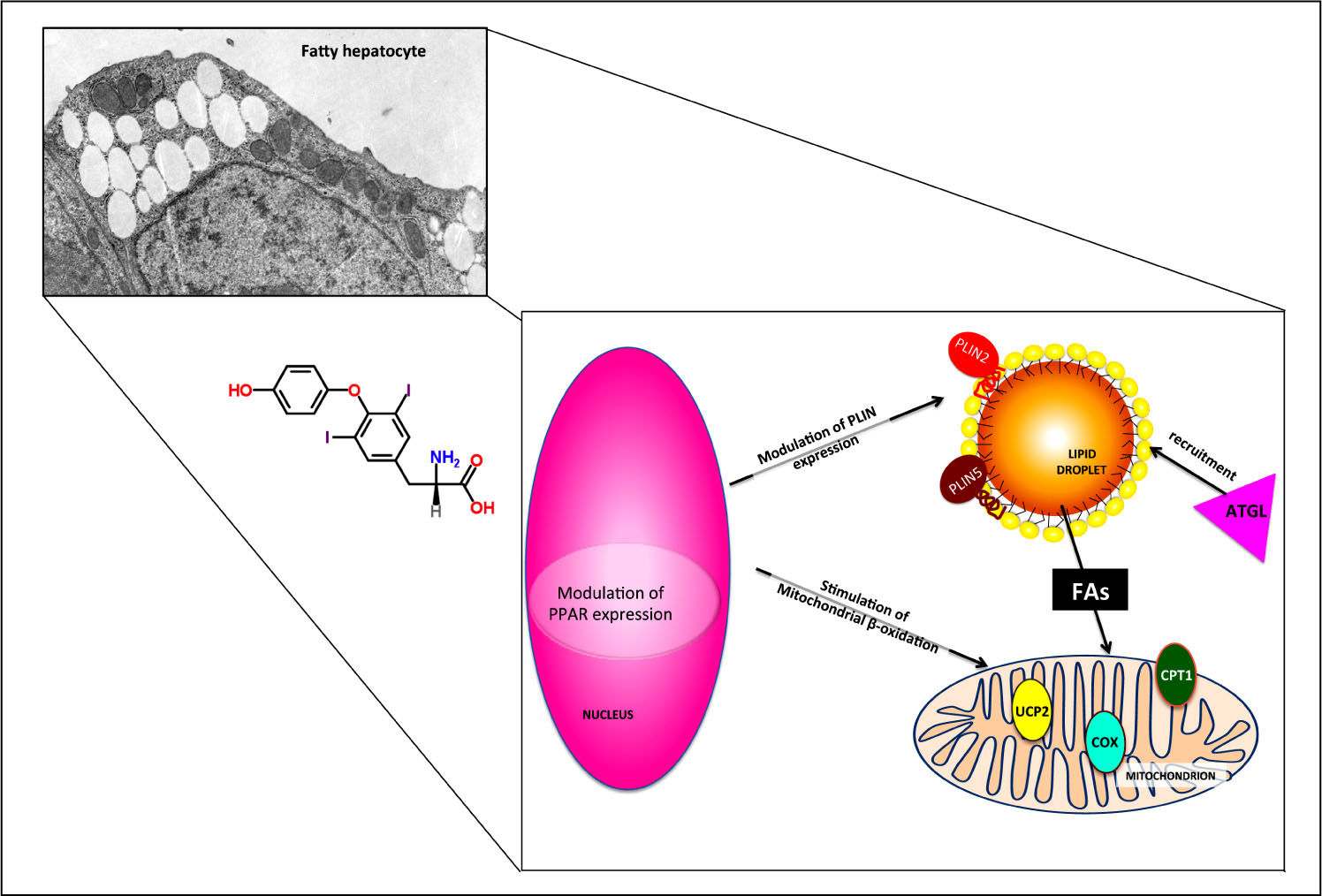
The use of primary rat hepatocytes allowed to verify that the lipid-lowering effect of T2 was a direct action on the hepatocyte, but the involvement of thyroid hormone receptors (TRs) in mediating this action remained to be elucidated. Therefore, the same experiments were performed in the FaO rat hepatoma cell line, that is defective for functional TRs.68,70 Treatment of steatotic FaO cells with T2 for 24 h reduced both TAG content and the number and size of LDs (Figure 6A). As in primary cultured hepatocytes, T2 stimulated mitochondrial FA oxidation by up-regulating the expression of Cpt1 (Figure 6B). The lipid-lowering action of T2 was accompanied by a down-regulation of Pparγ expression and an up-regulation of Plin2 (Figure 6C-6D). Moreover, expression of Atgl, Plin5 and uncoupling protein 2 (Ucp2) was also up-regulated by T2.70 The possible role of these proteins in the mechanism underlying the lipid-lowering effects of T2 are schematically depicted in figure 7.
 is recruited on LD surface, thus inducing FA release form LD core and their subsequent channeling into mitochondrion. The up-regulation of CPT1 and UCP2 expression and the stimulation of cytochrome-c oxidase (COX) activity indicate a stimulation of mitochondrial function.")
Possible mechanisms underlying the direct lipid-lowering effects of T2. The effects of T2 on fatty hepato-cytes are partially TR-independent and occur both at the nuclear and cytoplas-mic level. PPAR and PLIN expressions are modulated and adipose triglyceride lipase (ATGL) is recruited on LD surface, thus inducing FA release form LD core and their subsequent channeling into mitochondrion. The up-regulation of CPT1 and UCP2 expression and the stimulation of cytochrome-c oxidase (COX) activity indicate a stimulation of mitochondrial function.
In conclusion, the use of the two in vitro models of fatty hepatocytes allowed to demonstrate that the lipid-lower-ing effects of T2 depend on a direct interaction with the hepatic cell that, at least in part, occurs via “non-receptor-mediated” mechanisms and involves a short-term action by direct stimulation of mitochondrial activity. Altogether, our data support the possible utilization of T2 as a pharmacological tool in the treatment of NAFLD, also in light of its lack of thyrotoxic effects.80
Potential Translational Value of Such StudiesMany efforts have been made in order to introduce reliable and simple models resembling the most important features of NAFLD. In vitro models have the advantage of reducing the use of animals by providing a simpler and highly reproducible system, where the mechanisms can be studied directly at the cellular level, keeping the translational value of the observed results. Therefore, in vitro models offer the possibility to test the therapeutic potential and elucidate the mechanisms of action of a large spectrum of synthetic and natural compounds, without exposing humans to unnecessary risks.81 All together our studies demonstrated the efficacy of T2 as anti-steatotic and hepatoprotective compound in both in in vivo and in vitro models of NAFLD. These models, especially in vitro model using FaO cell line, may be employed to test the therapeutic potential of other emerging compounds.
Conclusions and Future PerspectivesNAFLD is closely related to obesity and insulin resistance and can progress to the necro-inflammatory form of non-alcoholic steatohepatitis. The incidence of NAFLD is rapidly increasing in industrialized countries, and is steering the increased risk of cardiovascular disease and type 2 diabetes. It is therefore mandatory to identify healthier lifestyles and to target NAFLD and its progression by highly active molecules.82 Whereas human studies are often burdened by several ethical, economical and time limitations, both animal and cellular models still provide valuable information in this field and will be confirmed at a translational level by clinical studies.47,83In vitro approaches can be particularly useful to study the molecular mechanisms involved in the onset and progression of NAFLD and to check the effects and mechanisms of action of endogenous and exogenous compounds, as shown in the case of T2.
Competing InterestThe authors declare that they have no competing interests.
AcknowledgmentsWe wish to thank for their collaboration Dr. Rita De Matteis and Mr. Valter Capicchioni. Many thanks to Prof. Gabriella Gallo for encouragements and stimulating discussions. Funding support for the studies mentioned here came from MIUR-COFIN 2008 (Prot. 20089SR S2X_002); Compagnia San Paolo, Torino (prat. no. 2009.1824-1067/IT/ pv); Fondi Ateneo Universitá degli Studi di Genova, Area-05, and Fondazione CARIGE (Prot. no. 2010/584-23), and European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 722619.



