




The role of nonalcoholic fatty liver disease, namely nonalcoholic steatohepatitis (NASH), as risk factor for liver- and non-liver-related morbidity and mortality has been extensively reported. In addition to lifestyle changes, capable of removing the metabolic factors driving disease progression, there is an urgent need for drugs able to reduce hepatic necroinflammation without worsening of fibrosis. This goal is considered by regulatory agencies as surrogate marker to define the effectiveness in pharmacological compounds in NASH, and fast-track approval was granted by the Food and Drug Administration in consideration of disease severity and unmet medical needs. Several compounds are in the pipeline of pharmaceutical industries and are being studied in phase II trials, but only a few (obeticholic acid, elafibranor) have started phase III trials. This concise review is intended to offer a systematic analysis of the most promising therapeutic intervention in NASH. In conclusion, there is reasonable expectation that drug may help curb the burden of NASH, and we look forward to obtaining solid data on their long-term safety and effectiveness. However, we should not forget that behavioral interventions remain a mandatory background treatment, able to stop disease progression in compliant overweight/ obese patients, with results that compare favorably with - and add to - the beneficial effects of drug treatment.
Nonalcoholic fatty liver disease (NAFLD), defined by the presence of hepatic accumulation of triglycerides in the hepatocytes in the absence of any other etiology of liver disease, is the most common cause of chronic liver disease in the Western world.1 Its clinical-histologic phenotype extends from nonalcoholic fatty liver to nonalcoholic steatohepatitis (NASH), characterized by inflammation and progressive fibrosis, leading to cirrhosis2 and end stage liver disease,3 as well as to hepatocellular carcinoma.4Whereas the estimated prevalence of NAFLD ranges from 6% to 33% (median 20%) in the general population, also varying on the basis of diagnostic methods, the prevalence of NASH only ranges from 3 to 5%,1 but NASH-related cirrhosis has become the second leading indication for liver transplantation in the United States5 and its importance is also raising in Europe.6
NAFLD is highly associated with metabolic disorders, including obesity and type 2 diabetes mellitus and it is considered the hepatic expression of metabolic syndrome.7 As such, NASH is also associated with an increased risk of cardiovascular disease and cardiovascular mortality8 and of type 2 diabetes,9 and the role of NASH as the driver for non-liver-related morbidity and mortality has recently been reconsidered.10 Accordingly, several trials have tested the possible effects of pharmacological compounds to clear fat from the liver, to reduce or halt disease progression or, possibly, to revert fibrosis and cirrhosis. These studies are hampered by several difficulties, summarized in table 1.
Difficulties in therapeutic trials in NASH patients.
| Disease side: | Lack of positive diagnostic criteria. Uncertainty of surrogate markers. Slow disease progression. High prevalence, but different phenotypes. |
| Patients’ side: | Young age and job constraints. Scarce awareness of risk. Need for diagnostic liver biopsy. |
| Physicians’ side: | Scarce awareness of disease progression. Poorly defined therapeutic targets. Need for an integrated approach. |
In summary, uncertainties in patients’ diagnosis and disease stratification, the need of repeated liver biopsies in relatively young, free living subjects with job constraints, and the long duration of disease are all conditions limiting the acceptance of strict rules by most patients. These constraints may be overcome by patients’ careful selection in tertiary centers,11 but exclude the vast majority of cases observed outside liver units or simply in primary care. Similarly, a joint agreement from an American Association for the Study of Liver Diseases (AASLD)-U.S. Food and Drug Administration (FDA) Joint Workshop is providing some clues to select primary outcomes in NASH studies.12 The most commonly accepted primary outcome of NASH studies is now set at reduction of NAS (NAFLD activity score), without worsening of fibrosis, or NASH reversal without worsening of fibrosis. These outcomes have been selected on the basis of the primary role of necro-inflammation and fibrosis as main drivers of disease progression.
Despite intensive research and numerous trials, as of today there are no approved treatments for NAFLD/NASH. The most favorable results have been obtained with the use of pioglitazone and/or vitamin E, whereas the effectiveness of other compounds, namely high-dose urso-deoxycholic acid, has never been systematically proven.13 In addition, there are no data on the long-term safety of these compounds.13
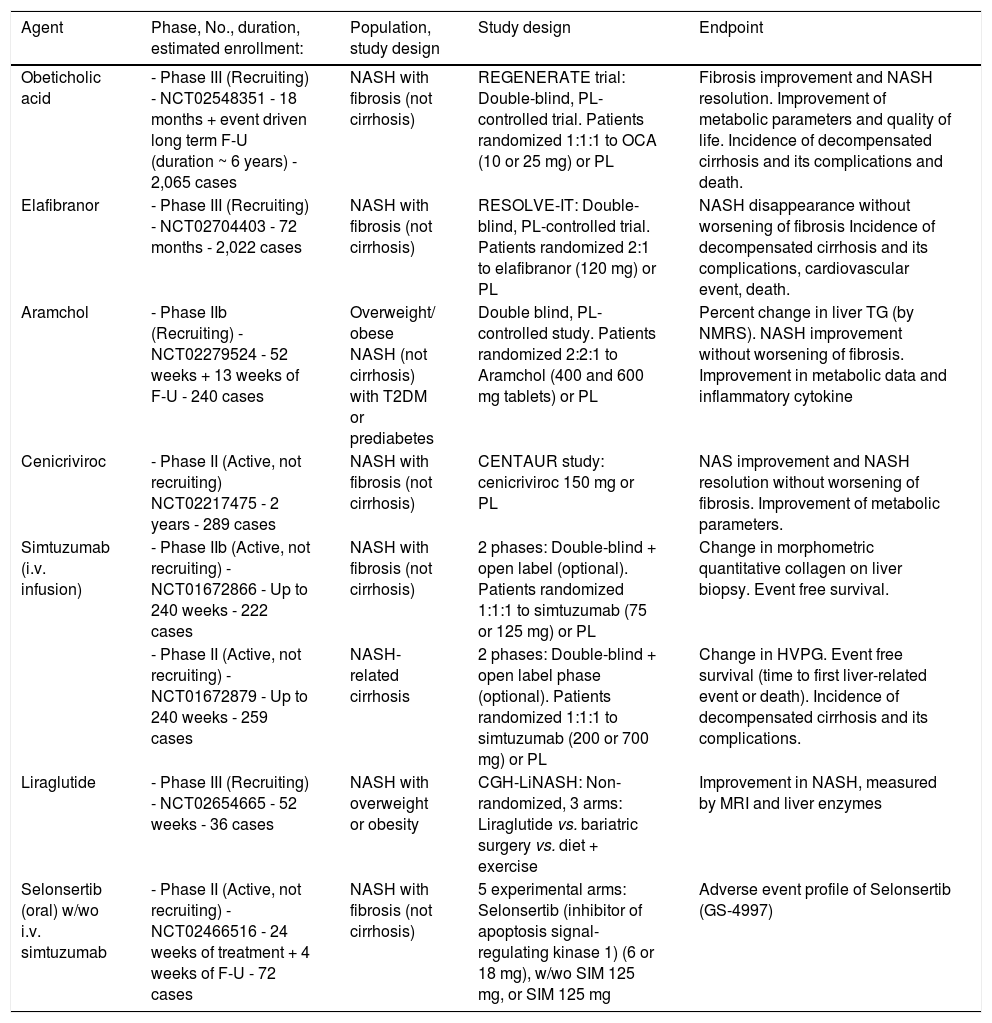
More recently, a series of drugs have reached evidence of beneficial effects in phase II studies (see below) and are being studied in long-term phase III studies (Table 2). These drugs offer several advantages in selected populations, and the results are reviewed in the next session.
Summary of ongoing randomized clinical trials for the treatment of NAFLD/NASH, as present in http://www.Clinicaltrials.gov
| Agent | Phase, No., duration, estimated enrollment: | Population, study design | Study design | Endpoint |
|---|---|---|---|---|
| Obeticholic acid | - Phase III (Recruiting) - NCT02548351 - 18 months + event driven long term F-U (duration ~ 6 years) - 2,065 cases | NASH with fibrosis (not cirrhosis) | REGENERATE trial: Double-blind, PL-controlled trial. Patients randomized 1:1:1 to OCA (10 or 25 mg) or PL | Fibrosis improvement and NASH resolution. Improvement of metabolic parameters and quality of life. Incidence of decompensated cirrhosis and its complications and death. |
| Elafibranor | - Phase III (Recruiting) - NCT02704403 - 72 months - 2,022 cases | NASH with fibrosis (not cirrhosis) | RESOLVE-IT: Double-blind, PL-controlled trial. Patients randomized 2:1 to elafibranor (120 mg) or PL | NASH disappearance without worsening of fibrosis Incidence of decompensated cirrhosis and its complications, cardiovascular event, death. |
| Aramchol | - Phase IIb (Recruiting) - NCT02279524 - 52 weeks + 13 weeks of F-U - 240 cases | Overweight/ obese NASH (not cirrhosis) with T2DM or prediabetes | Double blind, PL-controlled study. Patients randomized 2:2:1 to Aramchol (400 and 600 mg tablets) or PL | Percent change in liver TG (by NMRS). NASH improvement without worsening of fibrosis. Improvement in metabolic data and inflammatory cytokine |
| Cenicriviroc | - Phase II (Active, not recruiting) NCT02217475 - 2 years - 289 cases | NASH with fibrosis (not cirrhosis) | CENTAUR study: cenicriviroc 150 mg or PL | NAS improvement and NASH resolution without worsening of fibrosis. Improvement of metabolic parameters. |
| Simtuzumab (i.v. infusion) | - Phase IIb (Active, not recruiting) - NCT01672866 - Up to 240 weeks - 222 cases | NASH with fibrosis (not cirrhosis) | 2 phases: Double-blind + open label (optional). Patients randomized 1:1:1 to simtuzumab (75 or 125 mg) or PL | Change in morphometric quantitative collagen on liver biopsy. Event free survival. |
| - Phase II (Active, not recruiting) - NCT01672879 - Up to 240 weeks - 259 cases | NASH-related cirrhosis | 2 phases: Double-blind + open label phase (optional). Patients randomized 1:1:1 to simtuzumab (200 or 700 mg) or PL | Change in HVPG. Event free survival (time to first liver-related event or death). Incidence of decompensated cirrhosis and its complications. | |
| Liraglutide | - Phase III (Recruiting) - NCT02654665 - 52 weeks - 36 cases | NASH with overweight or obesity | CGH-LiNASH: Non-randomized, 3 arms: Liraglutide vs. bariatric surgery vs. diet + exercise | Improvement in NASH, measured by MRI and liver enzymes |
| Selonsertib (oral) w/wo i.v. simtuzumab | - Phase II (Active, not recruiting) - NCT02466516 - 24 weeks of treatment + 4 weeks of F-U - 72 cases | NASH with fibrosis (not cirrhosis) | 5 experimental arms: Selonsertib (inhibitor of apoptosis signal-regulating kinase 1) (6 or 18 mg), w/wo SIM 125 mg, or SIM 125 mg | Adverse event profile of Selonsertib (GS-4997) |
F-U: follow up. HVPG: hepatic vein pressure gradient NAS: NASH activity score. NMRS: nuclear magnetic resonance spectroscopy. MRI: magnetic resonance imaging. OCA: obeticholic acid. PL: placebo. SIM: simtuzumab. T2DM: type 2 diabetes mellitus. TG: triglycerides.
Liraglutide is a long-acting human GLP-1 analogue, which improves pancreatic beta-cell function by inducing insulin secretion while reducing glucagon release in a glucose-dependent manner. 14 In addition, liraglutide reduces appetite and delays gastric emptying, resulting in weight loss.15 Since 2006, Extendin-4, the first GLP-1 analogue discovered, and later liraglutide, have been demonstrated to reverse hepatic steatosis in experimental animals.16–18This activity might depend on the effect of GLP-1 analogues on body weight and systemic insulin resistance, although studies have also reported that these analogues can act directly on human hepatocytes in vitro, reducing steatosis by decreasing de novo lipogenesis and increasing fatty acid oxidation.16,19,20
Very recently, the safety and the efficacy of liraglutide in the treatment of NASH have been compared in a phase II randomized trial on 52 overweight subjects with and without diabetes (LEAN study), at the dose of 1.8 mg/day for 48 weeks.21 Nine of 23 patients who received liraglutide (39%) had resolution of definite NASH at the end-of-treatment liver biopsy, compared with two (9%) of 22 patients in the placebo group (relative risk (RR), 4·3 [95% CI 1.0-17.7]; p = 0.019). Only 2 cases in the liraglutide group vs. eight in placebo had progression of fibrosis (RR, 0.2 [0.1-1.0]; p = 0.04). As to safety, gastrointestinal side-effects were observed in 81% of cases on liraglutide and 65% on placebo; they included diarrhea (38% vs. 19%), constipation (27% vs. 0), and loss of appetite (31% vs. 8%). The authors conclude that liraglutide is safe, well tolerated and potentially useful to achieve histological resolution of non-alcoholic steatohepatitis, although data need confirmation in larger and long-term studies. Notably, following the SCALE study, liraglutide is one of the very few drugs approved for weight loss in the U.S. and in the European countries,22 and has been recently reported to reduce cardiovascular risk in type 2 diabetes.23
Obeticholic acidObeticholic acid (OCA) is a modified bile acid derived from the primary bile acid cheno-deoxycholic acid (CDCA). For many years, the physiological effects of bile acids were related to their physicochemical properties; more recently it was reported that bile acids also act as signaling molecules regulating not only their own homeostasis via their enterohepatic circulation.24 CDCA is the natural ligand for farnesoid X receptor (FXR), a nuclear receptor expressed at high levels in the liver, kidney, intestine and adrenal glands. In the liver, FXR is expressed in the hepatocytes, endothelial cells and Kupffer cells and, at a low level, in hepatic stellate cells.25 Nuclear receptors constitute a family of ligand-activated transcription factors that can either activate or repress a multitude of target genes.
OCA is 100-fold more potent than the endogenous FXR agonist CDCA, which makes OCA an attractive new therapeutic agent for NAFLD/NASH due to its multiple FXR-mediated effects. OCA increases glucose-stimulated insulin secretion, reduces blood glucose by enhancing peripheral glucose uptake, and inhibits hepatic lipid synthesis and content while inducing lipid uptake by adipocytes. FXR-mediated hepatoprotective properties of OCA include hepatocyte protection against bile acid-induced cytotoxicity, anti-inflammatory effects in liver and vasculature, and prevention and/or reversal of liver fibrosis.26,27 Thus, there is a strong rationale to promote OCA for the treatment of NASH.
Nonclinical studies have shown several potentially beneficial properties of FXR agonism in NASH, in particular FXR controls glucose metabolism through regulation of gluconeogenesis and glycogenolysis in the liver, as well as regulation of peripheral insulin sensitivity in striated muscle and adipose tissue,28,29 whereas the absence of endogenous intact FXR signaling results in dyslipidemia and a hepatic phenotype similar to NASH patients.30 On the contrary, FXR agonists reduce plasma triglycerides by repressing hepatic sterol regulatory element binding protein 1-c31 and increased hepatic fatty acid oxidation.32 In primary and cultured hepatocytes treated with pro-inflammatory mediators, OCA exerts direct inhibitory effects on pro-inflammatory gene expression.33 Based on pre-clinical evidence, OCA is under investigation for the treatment of multiple chronic liver diseases in humans, including the treatment of NASH, primary sclerosing cholangitis (PSC), biliary atresia, primary biliary cholangitis34 and other chronic liver diseases, as well as type 2 diabetes and NAFLD, where it demonstrated an insulin sensitizing effect.28
In the phase II FXR ligand OCA in NASH treatment (FLINT) study, OCA proved to be superior to placebo in improving most histologic features of the disease (inflammation, steatosis, ballooning and, in particular, fibrosis), as well as liver enzymes, also reducing weight and systolic blood pressure.35 However, low-density lipoprotein (LDL) cholesterol increased significantly in subjects treated with OCA compared to placebo (a modest increase well-responding to statin therapy), but treatment was accompanied by a relatively high rate of pruritus (23% OCA vs. 6% placebo), causing discontinuation in one case. A long-term phase III study is ongoing, and the results will be available in 2021.
ElafibranorElafibranor is a double peroxisome proliferator-activated receptor (PPAR) α/δ agonist, acting on nuclear receptors playing key roles in regulating metabolic homeostasis and inflammation.
PPARa are markedly expressed in the liver and can be activated by fibrates. Their activation results in increased uptake and oxidation of FFAs, increased triglyceride hydrolysis and upregluation of apolipoprotein (Apo)A-I and ApoA-II. The net effect is fatty acid oxidation, decrease in serum triglycerides, a rise in high-density lipoprotein cholesterol (HDL-C) levels, and an increase in cholesterol efflux. PPARa activation has also anti-inflammatory effects via inhibition of cyclooxygenase-2, interleukin-6, and C-reactive protein.36 PPARδ are widely expressed on fat, liver, skeletal muscle and heart tissues; their activation increases fatty acid transport and oxidation, improves insulin sensitivity and inhibits hepatic glucose output,37 and regulate macrophage inflammatory responses. Considering the emerging role of Kupffer cells in the pathogenesis of NAFLD, PPARδ might be crucial signaling receptors, also controlling the phenotypic switch between classical pro-inflammatory and alternative anti-inflammatory (M2) macrophages.38
Treatment with elafibranor in a mouse model of dyslipidemia has been demonstrated to lower both plasma triglycerides and total cholesterol and to increase plasma HDL-C levels.39 In phase IIa trials in dyslipidemic, prediabetic and type 2 diabetic patients, elafibranor ameliorated plasmatic lipid profile and glucose homeostasis, hepatic and peripheral insulin resistance and reduced liver inflammatory markers.40,41
Very recently, the efficacy and safety of elafibranor at 80 and 120 mg QD for 52 weeks have been tested in a phase IIb placebo controlled trial (GOLDEN 505) in 276 NASH patients.42 Elafibranor was well tolerated, at both doses; at a dose of 120 mg, elafibranor was effective on NASH resolution without worsening of fibrosis in patients with an active disease (NAS ≥ 4). Notably, elafibranor at 120 mg also improved the cardiometabolic risk profile of NASH patients by reducing plasma triglycerides, total and LDL-C, increasing HDL-C and improving glucose homeostasis, insulin resistance and inflammation. Clinical adverse events were generally mild to moderate in severity: cutaneous rash, decrease in appetite, arthralgia, dizziness and renal impairment were reported only in the elafibranor-treated arms. There were no differences in the number of severe adverse events between treatment and placebo groups.
In conclusion, clinical data confirm the good safety profile and the positive effect of elafibranor in NASH patients, with efficacy on histology associated with improvement on insulin resistance, reduced markers of liver cell necrosis (aminotransferases and γ-glutamyl-transpeptidase), and reduced inflammatory markers. A much larger phase III study is recruiting.
GliflozinsSodium-glucose co-transporter 2 (SGLT2) inhibitors reduce glucose reabsorption in the proximal tubule, thus reducing plasma glucose levels. Their use is associated with a moderate reduction of body weight, probably due to caloric loss related to glycosuria.43 A recent clinical trial revealed that empagliflozin treatment is associated with reduced cardiovascular mortality and maintained kidney function in patients with diabetes,44,45 which may be of additional value in subjects with NAFLD/NASH and the metabolic syndrome. Interest has risen from animal studies observing an effect of gliflozins on liver function in high-fat diet-induced obese rats and in choline-deficient l-amino acid-defined diet rats.46,47 Treatment with canagliflozin48 or dapagliflozin49 in subjects with diabetes decreased plasma aminotransferases, although there are no studies proving their effect on liver histology. Also remogliflozin has been shown to significantly improve markers associated with NAFLD in animal models, and may be a helpful compound for the treatment of NASH and NAFLD due to its specific insulin-sensitizing and antioxidant properties.50 In humans it has been shown to reduce HbA1c and to improve insulin sensitivity in subjects with type 2 diabetes. Post-hoc analysis in a 12-week trial in diabetes showed an approximate 40% reduction in ALT levels in subjects with elevated aminotransferase levels at baseline.51 These findings open a window for the therapeutic potential of gliflozins in NAFLD patients.
Simtuzumab (GS 6624)Simtuzumab is a monoclonal antibody against the enzyme lysyl oxidase-like 2 (LOXL2), responsible for the cross-linking of collagen and overexpressed during the progression of liver fibrosis. In animal models it has been shown that inhibition of LOXL2 results in a marked reduction in activated fibroblasts, desmoplasia and endothelial cells, reduced production of growth factors and cytokines and decreased signaling of transforming growth factor-beta (TGF-beta) pathway.52 When administered as subcutaneous injections in a phase II study on 20 patients, it was generally well-tolerated; the reported adverse events were abdominal pain, musculoskeletal pain, fatigue, and headache.52 Phase IIb studies in patients with advanced fibrosis secondary to NASH (with/without cirrhosis) are in progress.
CenicrivirocCenicriviroc is an oral, potent, dual antagonist of chemokyne receptor-2 and 5 (CCR2/CCR5). It demonstrated potent anti-inflammatory and anti-fibrotic activity in animal models of liver diseases.53,54 In humans, it significantly decreased the aspartate-to-platelet ratio index (APRI), noninvasive hepatic fibrosis index (FIB-4), and enhanced liver fibrosis score.55 It was well tolerated in a phase I study on 31 patients with mild or moderate hepatic impairment.56 Headache and gastrointestinal disorders (dry mouth, epigastric discomfort, flatulence) were the most commonly reported adverse events, but were of mild severity. Two phase II studies in patients with NAFLD are also under way.
AramcholAramchol (3b-arachidyl-amido, 7a-12a-dihydroxy, 5b-cholan-24-oic acid) is a synthetic lipid molecule obtained by combining 2 natural components, cholic acid (bile acid) and arachidic acid (saturated fatty acid), through a stable amide bond. Aramchol significantly reduces hepatic fat content in animals on a high-fat diet.57 In in vitro models, it achieves 70% to 83% inhibition of the stearoyl coenzyme A desaturase 1 (SCD1) activity, thus reducing the synthesis and increasing the β-oxidation of fatty acids, resulting in decreased hepatic accumulation of triglycerides and fatty acid esters.58 In addition, Aramchol activates cholesterol efflux by stimulating the adenosine triphosphate-binding cassette transporter A1, a pan-cellular cholesterol export pump,59 thus strengthening anti-atherogenic effects in animal studies. In preclinical studies, Aramchol at high doses did not cause the severe adverse effects attributed to complete inhibition of SCD1 (skin and eye disorders, inflammation, and atherosclerosis).60 In a phase IIb randomized, double-blind, trial of 60 patients with biopsy-confirmed NAFLD (six with NASH), patients were given Aramchol (100 or 300 mg) or placebo once daily for 3 months.61 No serious or drug-related adverse events were observed in the 58 patients who completed the study. Over 3 months, liver fat content (MRS-assessment) decreased by 12–22% in patients given 300 mg/day Aramchol, but increased by 6–36% in the placebo group (P = 0.02). Liver fat content non-significantly decreased also in the 100-mg Aramchol group.61 A larger phase II trial in overweight/obese patients with pre-diabetes or T2DM and NASH is in progress.
Galectin 3-inhibitorsGalectins are a family of proteins with binding specificities for β-galactoside sugars. Galectin-3 has cross-linking and adhesive properties and is coded by the lectin, galactoside-binding, soluble, 3 gene. There is evidence that galectin-3 is required for transforming growth factor (TGF)-β mediated myofibroblast activation and matrix production, and its regulatory gene may thus become a target of directacting antifibrotic agents. Knockout mice for the lectin, galactoside-binding, soluble 3 gene are indeed resistant to liver fibrosis induced by a variety of toxins.62 Phase 1 studies of galectin-3 inhibitors have been completed (GR-MD02; NCT01899859) and the agent is being evaluated in phase II studies of patients with NASH and cirrhosis (NCT02462967) or advanced fibrosis (NCT02421094).
Transmembrane G protein-coupled receptor (TGR) 5 agonists and dual FXR/TGR5 agonistsTGR5 is a classic G-protein coupled cell surface receptor widely expressed in various tissues (liver, gallbladder, bile ducts, adipose tissue, spleen, intestines, and kidneys); within the liver, TGR5 is abundantly expressed in Kupffer and endothelial cells, not in hepatocytes. TGR5 regulates the bile acid pool, modulates immune responses and increases energy expenditure (with significant effects on obese animals). Treatment of high-fat fed mice with a specific agonist (INT-777) reduced steatosis, improved liver enzyme levels without evidence of hepatic fibrosis, and improved insulin sensitivity.63 A dual FXR/TGR5 agonist (INT-767) is similarly effective, and improved the histological features of NASH in obese mice, also modulating cytokine production.64 Studies in humans are warranted.
The Next Future of Nash Drug DevelopmentThe clinical scenario of NAFLD/NASH treatment sees an impressive series of studies, either planned or in progress. A search on http://www.clinicaltrials.gov on July 24, 2016 identified 201 registered trials under the heading “NAFLD AND NASH AND Fatty Liver”, with more than 50% active at various stages of completeness, and this is definitely an underestimate of the total number of studies around the world. They reflect a specific interest of pharmaceutical companies for NAFLD/NASH, considering its epidemiology, of the medical societies because of the increasing awareness of its potential severity and complications, and of the regulatory agencies, considering the present and future burden of disease for the healthcare systems. Hoverer, there are specific challenges in regulatory trial design, in outcome research and in approval of drugs for this population, hampered by lack of surrogate endpoints, unknown disease modifiers, and the long way to valid end-points. These problems have been systematically reported in the joint AASLD-FDA workshop.12 In most cases, data will only be available in the next decade, but FDA granted fast-track approval to drugs for the treatment of NASH, a designation for drugs a) intended for the treatment of serious or life-threatening diseases or conditions; and b) able to address unmet medical needs for the disease or condition.
Table 2 summarizes the most important ongoing phase IIb or phase III studies, whose results are likely to provide important clues to NASH treatment. Most are based on histology outcomes, verified by repeated biopsies. Notably, all have a placebo arm, considering that no drugs have so far been approved for NASH treatment.
In most cases, the protocols of pharmacologic interventions also include standard lifestyle measures, which are known to improve steatosis, necroinflammation, and also reduce fibrosis and disease progression per se, and are a sort of background treatment. Unfortunately, very few centers have the resources and the skills to fully exploit lifestyle changes in the NASH population.65 When tested using the same outcomes as suggested by regulatory agencies for drug approval, also weight loss may result in NASH regression in overweight/obese patients. In a large prospective study of lifestyle intervention in NASH, all patients who lost 10% or more of their initial weight had a systematic reduction of NAS at follow-up liver biopsy after one year, 90% had NASH resolution, and 45% had regression of fibrosis.66 These results compare favorably with those achieved by pharmacologic intervention, might substantially contribute to prevent or even to cure diabetes,67 and might be associated with reduced cardiovascular risk.8
ConclusionsThe major difficulty in NASH treatment is related to its peculiar population, as therapy is expected to target free-living, asymptomatic subjects who are rarely aware of their future risk. Overweight/obesity and/or the metabolic syndrome are rarely considered significant illnesses, unless diabetes is present, and motivation for weight loss is rare unless cardiovascular comorbidities are present. Liver health is not a significant motivation for weight loss in the majority of patients, and it is difficult to achieve long-term compliance to dietary restriction and habitual physical activity in subjects who do not perceive their condition as a disease.68 As in other non-communicable diseases, both patients and physicians are keener to rely on drugs,69 but also adherence and long-term compliance to drug treatment remain a problem.
Several issues of pharmacologic treatment are still unresolved. First, what should we treat? Steatosis may be easily reversed, but is a cause of necroinflammation; necroinflammation is likely to drive fibrosis; fibrosis is associated with NASH progression. Accordingly, most trials are intended to address necroinflammation, as the pathogenic mechanisms of fibrosis. Second, who should be treated? There is no evidence that established cirrhosis might be effectively reversed; this means that treatment should be provided at initial stages to reduce the future burden of disease. This opens the last, unresolved question: is treatment cost-effective? The severity of advanced disease suggests that treatment -to be indefinitely continued if pathogenic factors are not removed- might be cost-effective, but only in selected, progressive cases. There is a considerable risk to expose many subjects to potentially severe adverse events to prevent the risk that an undetermined number of patients reach the condition of end-stage liver disease. A lot of additional information is needed to solve this pivotal issue.



