




Introduction and aim. Hepatocellular carcinoma (HCC) is the most common primary malignant liver tumor. It is primarily caused by hepatic cirrhosis or chronic viral hepatitis. Hepatic carcinogenesis is associated with increased oxidative stress. Thus, the aim of our study was to assess expression of the genes involved in the homeostasis of oxidative stress in patients with HCC.
Material and methods. The study was performed on 32 patients with primary HCC (verified by liver histology in 29 patients) and 27 control subjects (in 11 subjects, liver histology was available either with no or minimal changes in the liver tissue). Gene expressions of heme oxygenase 1 (HMOX1), biliverdin reductase A/B (BLVRA/B), NADPH oxidase 2 (NOX2)and p22phox were analyzed in the liver and peripheral blood leukocytes (PBL) in the subjects.
Results. Compared to controls, almost a 3 times higher mRNA level of BLVRA was detected in livers of HCC patients (p = 0.002); while those of BLVRB as well as HMOX1 were unchanged (p > 0.05). In accord with these results in the liver tissue, BLVRA mRNA levels in PBL were also significantly increased in HCC patients (p = 0.012). mRNA levels of NOX2 and p22phox in the liver tissue, although higher in HCC patients, did not differ significantly compared to control subjects (p > 0.05). Nevertheless, NOX2 mRNA level in PBL was significantly higher in HCC patients (p = 0.003).
Conclusions.BLVRA mRNA levels in the liver as well as in PBL are significantly higher in HCC patients most likely as a feedback mechanism to control increased oxidative stress associated with HCC progression.
Hepatocellular carcinoma (HCC) is the most common primary malignant liver tumor. The global mortality rate is 694,000 cases per year.1 Worldwide, HCC is the fifth most common cancer in men and the seventh in women; representing the third most frequent cause of cancer-related death.2 The incidence of HCC has different geographical distributions; sub-Saharan Africa, China, Hong Kong and Taiwan being among those regions with the highest incidence rates of HCC (i.e., more than 15 cases per 100,000 population per year.3 Conversely, North and South America and most of Europe are among those countries with a lower incidence. However, in recent years, the incidence rates have increased even in these regions, and this trend is expected to continue.4
This malignant disease arises in patients with chronic liver disease, mostly at the stage of liver cirrhosis. Almost 90 percent of cases are due to underlying cirrhosis or chronic hepatitis B and C virus infections.5 Well-defined etiological agents for the development of HCC are aflatoxin and excessive alcohol intake. Also, non-alcoholic steatohepatitis due to obesity, metabolic syndrome, and diabetes contribute significantly to the incidence of HCC.6
As far as the risk factors are known, screening programs for the risk groups can be established with an aim to detect tumors in the early stages. However, according to the available data, only 30% of patients with HCC are diagnosed in the early stages, when curative treatment is still possible.7 The recommended method of surveillance of HCC is a liver ultrasound at 6-month intervals;8 having sensitivity of about 65-80%, and a specificity of almost 90%.9
A combination of liver ultrasound and serum a1-fetoprotein (AFP) had been recommended in the previous guidelines. However, even combinations of these procedures is not sufficiently sensitive or specific to be used as a surveillance assay. AFP is typically increased in advanced tumors,10 but can be elevated in cholangiocarcinoma, liver metastases of colorectal cancer, gastric, testicular, or ovarian cancer; and it is also raised in cirrhosis. At the time of diagnosis, over 30% of HCC patients have normal serum levels of AFP.11 According to current AASLD guidelines, AFP serology is still considered an inadequate screening test for HCC.
Thus, new biomarkers are needed for early diagnosis of HCC. In fact, several of them are now under investigation including oxidative stress markers, angiogenic growth factors, or other markers such as glypican-3,12 lectin-bound AFP or des-γ carboxyprotrombin.13 However, so far, none of these, has been adequately investigated to be recommended as a screening test.
Hepatic carcinogenesis is a complex, multi-step process involving all pro-oncogenic and protective mechanisms. Increased production of reactive oxygen (ROS) and nitrogen species (RONS) is considered to be a trigger point in hepatic carcinogenesis.
NADPH oxidase (NOX) is a multiprotein enzyme complex importantly involved in ROS production,14 a phenomenon believed to contribute significantly to the apoptosis of liver cells.15NOX2, NADPH oxidase prototypic isoform is activated by the p22phox protein, which stabilizes and binds it to other subunits.16
The important enzyme in the antioxidant defense is heme oxygenase (HMOX), having two isoforms - HMOX1, highly inducible by oxidative stress, and HMOX2, the constitutive isoenzyme.17HMOX catalyzes the degradation of heme to biliverdin, carbon monoxide, and iron. Although controversies exist on the role of HMOX1 in carcinogenesis,18 both biliverdin and carbon monoxide exert important protective effects against oxidative stress.17
Another key enzyme in the heme catabolic pathway is biliverdin reductase (BLVR), reducing biliverdin to bilirubin, believed to be the most potent endogenous antioxidant substance.19 BLVR exists in two isoforms - BLVRA, the major enzyme in adults, and BLVRB, the predominant isoform in in the fetus.20BLVRA has multiple additional functions also acting as a transcription factor,21 a unique serine/threonine/tyrosine kinase,22 as well as cell membrane receptor involved in the immune response.23 Its role in carcinogenesis still remains to be elucidated.24
The aim of our study was to assess the expressions of those genes involved in the homeostasis of oxidative stress in patients with HCC.
Material and MethodsSubjectsThe study was performed on 32 patients with primary HCC (verified by liver histology in 29 patients) and 38 control subjects. The HCC patients were diagnosed, followed, and treated in the Military University Hospital in Prague between 2011 - 2014. Diagnosis of HCC was made by clinical, laboratory, and imaging (CT, MRI) examination. Liver histology was available from 29 patients (23 from CT-guided biopsies, in the remaining 6 patients the material was obtained from surgically-resected tissue).
Blood samples were analyzed in 32 patients with HCC - those with a verified diagnosis by histological examination; plus those with a likely diagnosis of HCC without histological verification, but diagnosed radiologically (typical imaging features were present in a contrast-enhanced study via dynamic CT-scan or MRI). A liver biopsy was not performed in these patients due to disapproval of the patient, advanced stage of the disease or contraindication of a liver biopsy.
As controls, 27 healthy volunteers (blood donors or employees of General Faculty Hospital and 1st Faculty of Medicine, Charles University in Prague) were used for gene expression studies in PBL. Eleven subjects who underwent a liver biopsy which resulted in no or minimal changes in the liver tissue (5 with non-alcoholic fatty liver disease, 3 with minimal changes, and 3 with normal liver histology) were used as controls for gene expression studies in liver tissue.
The study was registered under ID: NCT00842205 (www.clinicaltrial.gov). The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki. All subjects involved in the study had provided prior written informed consent.
Material sampling and storageLiver samples obtained from routinely performed CT-guided biopsies, or by liver tissue excision during surgical procedure were immediately placed into a RNAlater (Ambion Diagnostics, Austin, TX, USA) and stored at − 80°C. Blood samples for gene expression analyses were collected into PAXgene Blood RNA Tubes (PreAnalytix, Hombrechtikon, Switzerland) and stored at −80°C until total RNA isolation.
Total RNA isolation and reverse transcriptionHomogenization of liver tissue and isolation of total RNA was performed using RNeasy Mini (Qiagen, Dallas, TX, USA), isolation total RNA from PBL using a PAX-gene kit (Qiagen, Dallas, TX, USA), according to the manufacturer’s instructions. DNase treatment with RNase-free DNase (Qiagen, Dallas, TX, USA); prior to cDNA synthesis was carried out according to the manufacturer’s instructions. First-strand cDNA was synthesized from 0.2 µg of total RNA in a final volume of 20 µg using a High-Capacity cDNA kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions.
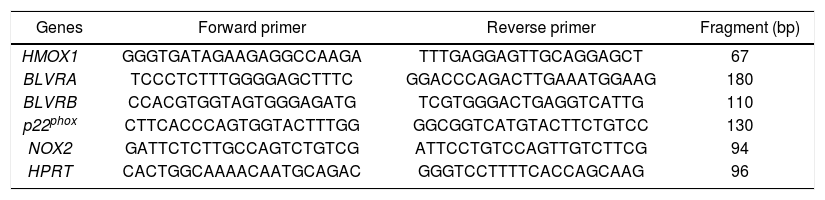
Gene expression quantificationThe BLVRA, BLVRB, HMOX1 and hypoxanthine phosphoribosyl transferase (HPRT) primer sequences were used as described previously.25 Primers for NOX2 and p22phox were designed using Primer 3 software (http://frodo.wi.mit.edu/primer3/, accessed 2013 Feb 01) and synthesized by Generi Biotech (Hradec Králové, Czech Republic) (Table 1).
Primer sequences for target and internal control genes.
| Genes | Forward primer | Reverse primer | Fragment (bp) |
|---|---|---|---|
| HMOX1 | GGGTGATAGAAGAGGCCAAGA | TTTGAGGAGTTGCAGGAGCT | 67 |
| BLVRA | TCCCTCTTTGGGGAGCTTTC | GGACCCAGACTTGAAATGGAAG | 180 |
| BLVRB | CCACGTGGTAGTGGGAGATG | TCGTGGGACTGAGGTCATTG | 110 |
| p22phox | CTTCACCCAGTGGTACTTTGG | GGCGGTCATGTACTTCTGTCC | 130 |
| NOX2 | GATTCTCTTGCCAGTCTGTCG | ATTCCTGTCCAGTTGTCTTCG | 94 |
| HPRT | CACTGGCAAAACAATGCAGAC | GGGTCCTTTTCACCAGCAAG | 96 |
HMOX1; heme oxygenase 1. BLVRA; biliverdin reductase A. BLVRB; biliverdin reductase B. NOX2: NADPH oxidase 2. HPRT: hypoxanthine phosphoribosyl transferase.
To determine the relative gene expression level of all data analysis, HPRT mRNA expressions were measured as internal controls. The fold change was calculated as 2-ΔΔct. The qPCR was performed in a 20 µL reaction volume, containing 4 µL of five-fold diluted cDNA template from a completed RT reaction, 1x SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA), and 200 nM (400 nM for BLVRB, 1000 nM for p22phox) of forward and reverse primers. All RT-PCR were set up in 96-well optical plates, and run on an ABI PRISM 7500 Sequence Detector System (Applied Biosystems, Foster City, CA, USA).
The cycling conditions included polymerase activation at 95°C for 10 min, followed with 40 cycles of 95°C for 15 s, and 60°C for 60 s. PCR products were subjected to a melting curve analysis. All samples were analyzed in triplicates. PCR efficiencies for target and housekeeping cDNA were 96 -105%.
Serum biochemistrySerum markers of liver injury (ALT, AST, GGT, ALP) and bilirubin were analyzed by routine assays on an automated analyzer (Cobas R8000 Modular analyzer, Roche Diagnostics GmbH, Mannheim, Germany).
Hematologic parameters were also analyzed on automated analyzers - INR on ACL500 (Instrumentation Laboratory, Bedford, Laboratory, Bedford, Massachusetts, USA); hemoglobin and platelets on a Sysmex XCE-5000 a XT-2000i (Sysmex Corporation, Kobe, Japan), respectively.
Statistical analysisDue to the non-normal distribution, data are described as median and IQ range. Differences between the studied groups were evaluated using the Mann-Whitney rank sum test. All analyses were performed with alpha set to 0.05.
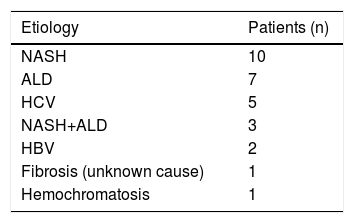
Results and DiscussionThe basic clinical and laboratory characteristics of our HCC patients are shown in table 2. The median age of our HCC patients was 69 years, HCC was almost 4 times more frequent in men than in women. The most prevalent underlying cause of HCC was non-alcoholic steatohepatitis (NASH), followed with alcoholic liver disease (ALD) (Table 3).
Clinical and laboratory characteristics of HCC patients.
| Gender (M:F ratio) | 3.83 |
|---|---|
| Age (years) | 69 (61.0 - 74.0) |
| Total bilirubin (µmol/l) | 15.3 (10.8 - 22.1) |
| ALT (µkat/l) | 0.53 (0.4 - 0.8) |
| AST (µkat/l) | 1.14 (0.8 - 1.6) |
| GGT (µkat/l) | 2.9 (1.2 - 6.2) |
| ALP (µkat/l) | 2.3 (1.7 - 4.4) |
| Albumin (g/l) | 33.9 ± 4.6 |
| INR | 1.22 (1.1 - 1.32) |
| Hemoglobin (g/l) | 121. 1 ± 19.1 |
| Platelets (× 109/l) | 203.5 ± 94.1 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline plhosphatase; GGT, gamma glutamyl transpeptidase; INR, international normalized ratio of prothrombin time. Data expressed as mean ± standard deviation, or median (IQ range) depending on data normality.
Hepatic carcinogenesis is a complex process, the understanding of which is still far from complete. Nevertheless, the role of increased oxidative stress and a dysfunctional antioxidant defense system seems to contribute significantly to the manifestation and progression of HCC (reviewed in reference 26). For instance, mice deficient in CuZn superoxide dismutase, which converts superoxide to H2O2, exhibit increased an incidence of HCC.27 Increased oxidative stress induced by hepatitis C virus infection, resulting in increased hepatic tumorigenesis, was also reported.28 The role of NOX, the major producer of superoxide in the mitochondria in mediating transforming growth factor (TGF)-β-induced hepatic fibrosis and carcinogenesis is also well recognized.29 In this context, it is interesting to note that bilirubin, one of the most important endogenous antioxidant substances,30 is a potent inhibitor of NOX.31,32
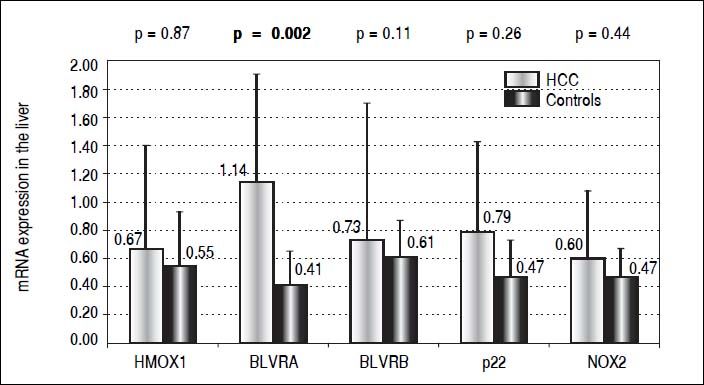
For decades the heme catabolic pathway has only been recognized only as a pathway required for the disposal of heme degradation products, but is now believed to play an important role in protection from increased oxidative stress.19 This pathway includes two important enzymes, HMOX and BLVR, reducing biliverdin to bilirubin, the major endogenous antioxidant. HMOX1, an inducible isoform, is a matter of controversy in terms of its role in carcinogenesis.18 While in some cancers HMOX1 gene expression may be viewed as a negative prognostic factor,33 clinical studies show that subjects with a more active HMOX1 gene variant are less likely to develop a variety of tumors (for review see reference 34). The protective role of HMOX1 was also reported in an animal model of hepatic carcinogenesis, demonstrating increased malignancy when HMOX1 was downregulated.35 However, in our study, we were not able to identify HMOX1 mRNA expression to be differentially modulated in HCC patients, either in the tumor tissue (0.67 ± 0.73 vs. 0.55 ± 0.38, p > 0.05) (Figure 1) or in PBL (1.91 ± 2.1 vs. 1.51 ± 0.62, p > 0.05). Nevertheless, BLVRA mRNA level was significantly upregulated in our HCC patients, both in tumor tissue and PBL. In fact, an almost 3 times higher mRNA levels of BLVRA were detected in livers of HCC patients compared to controls (1.14 ± 0.76 vs. 0.41 ± 0.24, p = 0.002). In accord with results in the liver tissue, BLVRA mRNA level in PBL was also significantly increased in our HCC patients (1.17 ± 0.46 vs. 0.90 ± 0.29, p = 0.012).

These results are in accord with recent data by De Giorgi, et al. on patients with HCV-induced HCC36 as well as our own results demonstrating increased BLVRA mRNA expression in HCV infected patients.25 Our data are also corroborated by the immunohistological study by Arena, et al., who showed increased protein expression of BLVR in tumor tissues of patients with melanoma.37 Overexpression of a BLVRA protein was also reported in clinical renal cancers,38 as well as vaginal carcinomas.39BLVRB, the other BLVR isoenzyme being predominantly important during fetal life, was reported to be upregulated on a protein level in HCC patients by Melle, et al.;40 and its possible pro-carcinogenic role in HCC was also described in a recent experimental study by Huan, et al.41 However, we were not able to confirm this data, since only a mild and non-significant elevation of BLVRB mRNA levels was found in our HCC patients compared to controls (0.73 ± 0.97 vs. 0.61 ± 0.26, p > 0.05) (Figure 1).
The functional significance of increased BLVRA mRNA expression remains to be answered. One explanation might be feedback stimulation of the antioxidant defense, which is what we believe is true in HCV-infected patients; those who responded to antiviral therapy had much higher BLVRA mRNA expression compared to non-responders.25 The beneficial role of BLVRA in preventing oxidative stress-induced senescence was also reported,42 supporting this hypothesis. On the other hand, BLVRA silencing in renal cells had a pro-apoptotic effect,43 and BLVRA, surprisingly serving as a transcription factor, is a known activator of multiple pro-proliferative intracellular signaling pathways.24BLVRA is also a sensor of intracellular hypoxia; indeed, its expression has been shown to be significantly increased in response to hypoxia.44 Thus, it seems that several mechanisms are behind the up-regulated BLVRA observed in biological studies.
Our explanation of increased BLVRA mRNA expression due to increased oxidative stress might be plausible, as evidenced by increased NOX2 mRNA levels in the PBL of our HCC patients. Although mRNA levels of NOX2 and p22phox in the liver tissue only showed a non-significantly higher trend in our HCC patients (0.60 ± vs. 0.47, and 0.79 ± 0.64 vs. 0.47 ± 0.26, respectively, p > 0.05 for both comparisons) (Figure 1), mRNA level of NOX2 in PBL was significantly higher in these HCC patients (1.91 ± 1.21 vs. 1.22 ± 0.52, p = 0.003). Thus, BLVRA may act as a feedback mechanism to scavenge superoxide overproduced by increased NOX2.26
It is also important to emphasize the importance of the PBL as a biological material to be used for screening expression studies. The PBL are easily available from blood sampling, and their gene expression profiles are more reliable compared to liver cancers often containing necrotic tissues.44
ConclusionIn conclusion, we observed increased BLVRA mRNA level in the liver as well as in PBL in HCC patients, which seems to be a feedback mechanism to control increased oxidative stress associated with HCC progression, as evidenced by increased NOX2 mRNA levels in PBL of these patients. We were not able to assess either the BLVRA protein levels or BLVRA enzyme activities in our biological samples; thus further studies aimed to deeper analyze BLVRA as a possible therapeutic target are certainly needed.
Abbreviations- •
AFP: a1-fetoprotein.
- •
ALD: alcoholic liver disease.
- •
ALT: alanine aminotransferase.
- •
AST: aspartate aminotransferase.
- •
ALP: alkaline phosphatase.
- •
BLVR: biliverdin reductase.
- •
BLVRA: biliverdin reductase A.
- •
BLVRB: biliverdin reductase B.
- •
GGT: gamma glutamyl transpeptidase.
- •
HBV: viral hepatitis B.
- •
HCC: hepatocellular carcinoma.
- •
HCV: viral hepatitis C.
- •
HMOX1: heme oxygenase 1.
- •
HPRT: hypoxanthine phosphoribosyl transferase.
- •
INR: international normalized ratio of prothrombin time.
- •
NASH: non-alcoholic steatohepatitis.
- •
NOX2: NADPH oxidase 2.
- •
PBL: peripheral blood leukocytes.
- •
RONS: reactive nitrogen species.
- •
ROS: reactive oxygen species.
- •
TGF: transforming growth factor.
This work was supported by grant IGA MZ NT 13092-4/2012 from the Czech Ministry of Health.



