




Immunoglobulin G4 associated cholangitis (IAC) is an autoimmune disease associated with autoimmune pancreatitis (AIP). It presents with clinical and radiographic findings similar to primary sclerosing cholangitis (PSC). IAC commonly has a faster, more progressive onset of symptoms and it is more common to see obstructive jaundice in IAC patients compared to those with PSC. One of the hallmarks of IAC is its responsiveness to steroid therapy. Current recommendations for treatment of AIP demonstrate excellent remission of the disease and associated symptoms with initiation of steroid therapy followed by steroid tapering. If untreated, it can progress to irreversible liver failure. This report describes a 59 year-old female with un-diagnosed IAC who previously had undergone a pancreaticoduodenectomy for a suspected pancreatic cancer and later developed liver failure from presumed PSC. The patient underwent an uncomplicated liver transplantation at our institution, but experienced allograft failure within five years due to progressive and irreversible bile duct injury. Radiology and histology suggested recurrence of PSC, but the diagnosis of IAC was suspected based on her past history and confirmed when IgG4 positive cells were found within the intrahepatic bile duct walls on a liver biopsy. A successful liver retransplantation was performed and the patient is currently on triple immunosuppressive therapy. Our experience in this case and review of the current literature regarding IAC management suggest that patients with suspected or recurrent PSC with atypical features including history of pancreatitis should undergo testing for IAC as this entity is highly responsive to steroid therapy.
Over the past decade autoimmune pancreatitis (AIP) has become a better understood and described entity. In 1995, Yoshida, et al. described a case of chronic pancreatitis suspected of being caused by an autoimmune mechanism.1 Subsequently, patients with suspected sclerosing pancreatitis were found to have elevated serum levels of IgG4 and increased IgG4-positive plasma cells in the pancreas, further supporting the role of this etiologic pathway for these patients.2 Infiltration of IgG4-positive plasma cells has been noted not only in the pancreas of AIP patients, but in other tissues and organs including the liver, bile ducts and periportal tissues.3 Kami-sawa and Okamoto proposed that AIP was a pancreatic manifestation of an entity they named IgG4-related sclerosing disease.4 This disease was noted to include IgG4 related AIP, sclerosing cho-langitis, inflammatory pseudotumor, cholecystitis and a number of other disease processes.5
Mendes, et al., at Mayo Clinic described a small percentage (9%) of primary sclerosing cholangitis (PSC) patients with elevated serum IgG4 that manifested an increased severity of liver disease and shorter time to transplant. They suggested that this subset of patients behaved similar to AIP with biliary strictures.6 In 2007, Björnsson, et al. conducted an extensive review of case reports and case series of sclerosing cholangitis associated with pancreatitis as well as IgG4 positive cholangitis. They proposed this new entity be named IgG4 associated cholangi-tis (IAC). IAC closely mimics PSC clinically and ra-diographically. It is often associated with AIP and demonstrates chronic inflammatory changes of the bile ducts including plasma cells that are positive for IgG4. Initiation of prednisolone 0.6 mg/kg/day followed by a steroid dose taper has been demonstrated to be effective in treating AIP and its associated symptoms and AIP relapse is responsive to readmi-nistration of dose-up steroids.7,8 Likewise, IAC often responds to steroid therapy.9
Case ReportA 54 year-old female was referred to our institution for progressive end-stage liver disease complicated by recurrent episodes of cholangitis, hepatic encephalopathy, jaundice, fatigue, peripheral edema and ascites. She was diagnosed with liver disease six years prior when her liver enzymes and bilirubin levels were noted to be elevated at the time of a routine physical examination. She had an extensive work-up for the cause of her liver dysfunction at an outside institution that included liver biopsy, laboratory testing and imaging. A computed tomography scan revealed a pancreatic head mass and she subsequently underwent a pancreaticoduodenectomy. Pathological examination of the specimen revealed chronic pancreatitis and no evidence of malignancy.
Her liver function continued to decline with recurrent episodes of cholangitis accompanied by dilated, abnormal appearing bile ducts on imaging consistent with PSC. She was evaluated, listed and underwent an uncomplicated orthotopic liver transplant in 2005. She experienced recurrent cho-langitis episodes with steadily rising bilirubins and transaminases three years after OLT. Over the following two years she developed progressive end-stage liver disease with recurrent cholangitis, bacteremia and sepsis requiring percutaneous drainage of intra-hepatic ducts and chronic use of broad spectrum antibiotics.
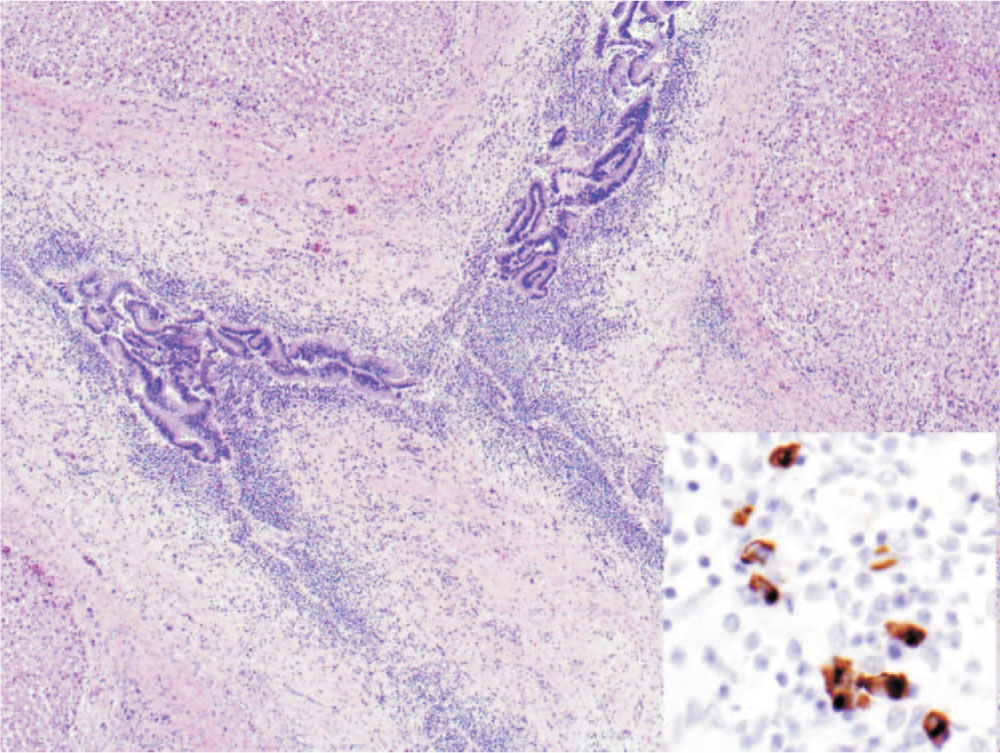
Given her history of previous pancreatitis, a benign pancreatic mass, recurrent cholangitis and imaging consistent with PSC, her native liver ex-plant was retrospectively immunostained for IgG4 to evaluate for possible IAC. The stain revealed numerous IgG4 positive plasma cells within the bile duct walls suggesting a diagnosis of IAC (Figure 1). It was concluded that the obstructing pancreatic mass removed by pancreaticoduodenectomy had most likely been an inflammatory mass of AIP.
. Inset shows numerous IgG4-positive plasma cells (immu-nostain, magnification x 400).")
As the patient’s condition continued to worsen, she was listed for liver retransplantation. She underwent a repeat OLT 11 years after her initial diagnosis of liver disease and 5 years after her first OLT. Pathologic examination of her explanted liver allograft revealed IgG4 positive plasma cells similar to those seen in her native liver supporting the diagnosis of IAC (Figure 2). She did well following her second OLT, with a transient rise in her liver enzymes treated by increasing her prednisone dose and not putting her on a standard prednisone taper that the majority of our patients are on.
Discussion. Inset shows scattered IgG4-positive plasma cells (immunostain, magnification x 400).")
The prevalence of IgG4 related disease is still not known. A 2003 study from Toronto, Canada over a 31 year period revealed that 7% of PSC patients had associated pancreatic findings.10 Similarly, a multi-center study covering 29 years and 388 PSC patients found a 7% association with AIP.11 This small, but significant subpopulation of PSC patients may represent a group a patients with significantly different therapeutic requirements and prognosis.
According to a recent literature, IAC occurs more frequently in men (2:1) and older patients compared to the average age of diagnosis for PSC. IAC can have a varied presentation, but often progresses at a faster rate and presents with obstructive jaundice compared to PSC. IAC and AIP patients will often have elevated serum IgG4 levels and increased IgG4 positive plasma cells on histologic examination of the inflamed tissue.6 Radiographically, segmental stenosis of the lower bile duct is observed in sclerosing cholangitis patients with AIP, but a beaded or pru-ned-tree appearance is seen only in PSC patients.12
AIP and IAC have been shown to respond remarkably well to steroid therapy.7-9 Given the significant long-term risk of liver failure with PSC and untreated IAC alike, it may be of benefit to screen atypical PSC patients and those with a history of pancreatic problems for elevated serum IgG4 levels. Likewise, if a patient progresses to liver disease with a diagnosis of PSC, it may be beneficial to examine the liver biopsy or explant for IgG4 positive plasma cells so that for immunosuppressive maintenance therapy can be optimized. As more data emer-ges on this disease process we may see all PSC patients being screened given the potential for effectively treating nearly a 10% of these patients with steroids. A number of case reports and case series of steroid treatment for IAC can be found in the literature; however no clear consensus for dosing and duration of steroid therapy specifically for IAC exists. Current recommendations for steroid treatment of AIP consists of an initial prednisolone dose of 0.6 mg/kg/day followed by a steroid dose taper with readministration of dose-up steroids for relapse.7,8 IAC that progresses to liver failure requiring liver transplant or, as in the case of our patient, IAC recurrence in a liver allograft may very likely necessitate a prolonged or lifelong steroid therapy in the post-transplant setting.



