











Acute liver injury is a current health problem with few effective treatments. The present study investigated the hepatoprotective and curative potential of the glucagon-like peptide-1 analog liraglutide against carbon tetrachloride (CCl4)-induced hepatotoxicity.
Materials and methodsMale Swiss mice were subjected to two protocols. The first protocol (Pretreatment) consisted of intraperitoneal (i.p.) treatment with liraglutide (0.057 and 0.118mgkg−1) or vehicle (distilled water) once daily for 7 days. On days 6 and 7, the animals were challenged with 2% CCl4 (5mgkg−1, i.p.). The second protocol (Late treatment) began with an injection of 5% CCl4 (5mgkg−1, i.p.) and subsequent treatment with liraglutide (0.057mgkg−1) or vehicle (distilled water) for 1 day. In both protocols, 24h after the last administration, blood and bile were collected from anesthetized animals, followed by euthanasia and liver collection. Plasma and bile underwent biochemical analyses, and histological, oxidative stress, and metabolic parameters were evaluated in the liver.
ResultsBoth liraglutide treatment protocols attenuated hepatotoxicity that was induced by CCl4, decreasing plasma levels of hepatic enzymes, stimulating the hepatic antioxidant system, and decreasing centrilobular necrosis, hepatic glycogen, and lipid accumulation. CCl4 tended to reduce bile lipid excretion, but liraglutide did not influence this parameter.
ConclusionsThe present results demonstrated the hepatoprotective and therapeutic effects of liraglutide, which may be attributable to a decrease in liver oxidative stress and the preservation of metabolism. Liraglutide may have potential as a complementary therapy for acute liver injury.
Intense or chronic exposure to pollutants, alcohol, and drugs can disrupt normal liver function [1]. Drug-induced liver injury can result in acute liver failure and is characterized as one of the most serious adverse effects of drugs. The incidence of drug-induced livery injury has steadily increased because of the substantial number of medications on the market and their abuse [2]. Hepatic damage that is promoted by drugs is directly associated with oxidative stress that results from excess free radicals that are generated through hepatic xenobiotic pathways [3].
Carbon tetrachloride (CCl4) is a toxic agent that is currently used as a well-established model of experimental hepatotoxicity based on its ability to promote centrilobular necrosis with fat deposition [4]. These alterations that are induced by CCl4 are attributable to trichloromethyl radicals (CCl3), reactive metabolites that are formed by hepatic metabolism via cytochrome P450 enzymes, mainly CYP2E1. Trichloromethyl radicals react rapidly with oxygen molecules, forming trichloromethyl peroxy radicals (OOCl3). Both are able to covalently bind to cellular structures, causing damage through oxidative stress and lipid peroxidation and consequently resulting in cellular death [5].
Therapeutic strategies for the treatment of acute liver injury are still limited. Removal of the causal agent and the use of antioxidant substances are the main treatment modalities. Several compounds have been studied for clinical application in these cases. Glucagon-like peptide-1 (GLP-1) analogs comprise a recent class of antidiabetic drugs that mimic the effects of incretins and have been reported to have beneficial effects in some organs. GLP-1 analogs have been shown to not cause changes in blood glucose when administered in non-diabetic patients, thus supporting their use for the treatment of other disorders [6]. Liraglutide is a long-acting synthetic analog of GLP-1 with 97% homology to human GLP-1. Liraglutide was shown to prevent oxidative stress and improve hepatic cell apoptosis in models of chronic liver disease [7] and hepatic glucolipotoxicity [8], respectively. The cytoprotective effects of liraglutide have been related to the activation of nuclear factor erythroid 2-related factor 2 (Nrf-2), a transcription factor that regulates the expression of redox homeostasis genes and is involved in the oxidative stress response and cellular defense mechanisms [9]. However, the involvement of GLP-1 analogs in acute liver injury is still unclear, and the therapeutic potential of liraglutide in such cases is still unclear. Most studies of liraglutide have only evaluated its preventive potential in chronic diseases. Thus, the present study investigated the protective and therapeutic effects of liraglutide on CCl4-induced hepatotoxicity and the underlying mechanisms and pathways that are involved in these actions.
2Material and methods2.1Hepatotoxicity induction and experimental designAdults male Swiss mice (Mus musculus), weighing 25±5g, were obtained from the vivarium of the Federal University of Paraná (Curitiba, Brazil). The Ethics Committee for Animal Care of SCB/UFPR approved all of the experimental protocols (approval no. 1101), that followed the National Institute of Health guidelines (USA). The animals were maintained under controlled room temperature (22±1°C) on a 12h/12h light/dark cycle with free access to food and water. Two different treatment protocols were employed to evaluate the protective and therapeutic potential of liraglutide against acute liver injury.
2.2Pretreatment with liraglutideThe animals were randomly divided into five groups (n=7–12/group) and treated with the compounds or water once daily for 7 days. On days 6 and 7 of treatment, the animals were challenged with CCl4 (2% in canola oil, 5mlkg−1, i.p.). The treated groups received liraglutide (0.057 and 0.118mgkg−1, i.p.) and injections of CCl4. Both doses of liraglutide were calculated by interspecific allometry [10] based on doses that are indicated for humans (0.6 and 1.2mg). The 0.057 and 0.118mgkg−1doses that were used in the present study are hereinafter referred to as the low dose (LD) and high dose (HD), respectively. The naive group received water and injections of canola oil. The vehicle group received water and was injected with CCl4. One additional positive control group was orally treated with N-acetylcysteine (NAC, 500mgkg−1) and injected with CCl4. Glycemia was monitored during the experiment using a glycosometer (AccuCheck®). Twenty-four hours after the last injection of CCl4, the mice were fasted for 12h and then intraperitoneally anesthetized with 100mgkg−1 ketamine and 10mgkg−1 xylazine. Blood samples were collected from the abdominal cava vein with heparinized syringes, and bile was collected from the gall bladder using ultra-fine insulin needles. Bile was stored at −20°C until analysis. After euthanasia, liver samples were immediately collected and stored at −80°C for further analysis. A portion of the liver was fixed in Alfac solution for histological analysis. The spleen, kidneys, and lungs were removed and weighed to evaluate possible macroscopic alterations. The experimental protocol is shown in Fig. 1A.
 or late treatment (B) according to the protocols that are described in Section 2.")
Experimental design in mice that were challenged with CCl4 and received liraglutide in pretreatment (A) or late treatment (B) according to the protocols that are described in Section 2.
The animals were divided into two groups (n=7–12/group) and challenged with a single dose of CCl4 (5% in canola oil, 5mlkg−1, i.p.). The treatment group received liraglutide (0.057mgkg−1, i.p.) or vehicle (distilled water) 1h after the CCl4 challenge. In this protocol, liraglutide was administered only at the LD. Glycemia was monitored during the experiment using a glycosometer (AccuCheck®). After 24h of treatment, the mice were intraperitoneally anesthetized with 100mgkg−1 ketamine and 10mgkg−1 xylazine, and blood, bile, and liver samples were collected as described in the first experimental protocol. The spleen, kidneys, and lungs were also removed and weighed. The experimental protocol is shown in Fig. 1B.
2.4Biochemical analysis of blood and bilePlasma was obtained by centrifuging the blood samples at 3400×g for 5min. Plasma was used to measure biochemical markers of liver function, including the activity of aspartate transaminase (AST), alanine transaminase (ALT), and alkaline phosphatase (ALP), using commercial kits in an automatic analyzer (Mindray BS-200, Shenzhen, China). Liraglutide is a hypoglycemic drug; therefore, plasma glucose levels were also measured during treatment. The bile was diluted in 0.9% saline solution for volume adjustment (100μl) and subsequently analyzed for total cholesterol in an automatic analyzer (Mindray BS-200, Shenzhen, China).
2.5Hepatic histopathologyHepatic tissue was rapidly harvested from the animals and fixed in Alfac (90% ethyl alcohol, 40% formaldehyde, and glacial acetic acid) for 24h. After being embedded in paraffin, 5μm sections were prepared, deparaffinized, and stained with hematoxylin and eosin (HE). The analysis was blindly performed using an optical microscope. Necrosis, inflammation, and cellular ballooning were evaluated. Lesions were scored according to the adapted Knodel system: 0 (no lesions), 1 (mild lesions with necrosis in ≤1/3 of tissue), 2 (moderate lesions with necrosis in 1/3–2/3 of tissue), and 3 (marked lesions with necrosis in ≥2/3 of tissue) [11].
2.6Determination of liraglutide antioxidant activity2.6.1Determination of hepatic oxidative stress parametersOxidative stress parameters were analyzed using hepatic tissue that was homogenized in potassium phosphate buffer. The pure homogenate was used to determine the levels of reduced glutathione – GSH [12] and lipoperoxidation – LPO [13]. The liver homogenate was then centrifuged at 21,000×g for 20min at 4°C, and the supernatant was used to measure the activity of hepatic glutathione S-transferase – GST [14], superoxide dismutase – SOD [15], and catalase – CAT [16]. With the exception of GSH, the results are expressed as the amount of protein in the liver samples, determined using the Bradford (1976) method [17]. All of the techniques were performed in 96-well microplates and read in a spectrophotometer (Synergy HT – Biotek, Winooski, VT, USA).
2.6.2Determination of free radical scavenging activity of liraglutideThe antioxidant potential of liraglutide per se was analyzed using the method of Chen et al. (2004), with modifications [18]. The technique consists of measuring the reactivity of liraglutide at different concentrations (1, 3, 10, 30, 100, 300, and 1000μgml−1) with the stable free radical 2,2-diphenyl-1-picrylhydrazyl (DPPH). Ascorbic acid solution (50μgml−1) was used as the positive control, and distilled water was used as the negative control. Absorbance was read in a microplate spectrophotometer before and 5min after the addition of DDPH.
2.7Hepatic glycogen determinationHepatic glycogen levels were measured according to Kepler and Decker (1974), with modifications [19]. Briefly, frozen hepatic samples were homogenized in 0.6N perchloric acid, and basal glucose levels were determined using a commercial kit (Labtest, Lagoa Santa, Brazil). Afterward, the homogenates underwent glycogen hydrolysis using 0.2M amyloglucosidase and maintained in a 40°C water bath for 60min. The reaction was stopped by the addition of 0.6N perchloric acid and centrifuged at 7600×g for 10min. The supernatants were used to determine the final glucose levels. Both assays were read at 505nm. Glycogen levels were calculated as the difference between the basal and final glucose levels. The results are expressed as μmolgtissue−1.
2.8Lactate and pyruvate analysisFrozen hepatic samples were homogenized in 0.6N perchloric acid as described above and centrifuged at 10,000×g for 10min. The supernatant was used to measure hepatic lactate and pyruvate levels [20,21]. Both metabolites were determined using standard enzymatic techniques based on NAD+ reduction and NADH oxidation, respectively. The microplates were read at 340nm. The results are expressed as μmolgtissue−1.
2.9Hepatic gravimetric analysisTissue gravimetry was performed to determine the influence of liraglutide on total liver lipid content according to Folch et al. (1957), with modifications [22]. Briefly, the liver samples were lyophilized and then mixed with hexane. The mixtures were heated to 80°C for 2h, and the supernatant was transferred to a second glass tube for natural evaporation. This procedure was repeated three times. The lipid content was weighed and suspended in 99.50% chloroform and 99.50% isopropanol for the determination of hepatic total cholesterol using commercial kits with an automatic analyzer. The results are expressed as mggtissue−1.
2.10Statistical analysisThe statistical analyses were performed using Prism 5.0 software (GraphPad, San Diego, CA, USA). The results are presented as the mean±SEM. Group differences were assessed using one-way analysis of variance (ANOVA) followed by the Bonferroni post hoc test or unpaired two-tailed Student's t-test. Values of p<0.05 were considered statistically significant.
3Results3.1Liraglutide attenuated hepatic alterationsHepatic damage that was caused by CCl4 was measured based on relative liver weight, and plasma hepatic biomarkers were quantitatively analyzed. All of the groups that were challenged with CCl4 presented an increase in relative liver weight compared with the vehicle group. Pretreatment with liraglutide at the HD (0.118mgkg−1) partially prevented the increase in relative liver weight (Fig. 2A), whereas this effect was not observed with the late treatment protocol (Fig. 2B). The weight of the spleen, kidneys, and lungs did not differ between groups (Supplementary Table S1). Increases in the activity of plasma hepatic enzymes (ALT, AST, and ALP) were detected in the groups that received CCl4, confirming tissue damage. The groups that were treated with liraglutide with both protocols exhibited significant reductions of ALT, AST, and ALP activity. Liraglutide pretreatment decreased plasma levels of ALT by approximately 39% (p=0.045) (Fig. 3A). Late treatment with liraglutide decreased plasma levels of ALT by 56% (p=0.021) (Fig. 3D). Similarly, plasma AST levels decreased by ∼41% (p=0.049) and 59% (p=0.033) with the pretreatment and late treatment protocols, respectively (Fig. 3B, E). The levels of ALP were slightly decreased by both treatment protocols, but these decreases were not statistically significant (Fig. 3C, G). The two doses of liraglutide presented similar effectiveness. Therefore, the LD (0.057mgkg−1) was used for the late treatment protocol and subsequent analyses. These results with liraglutide treatment were better than with N-acetylcysteine treatment (i.e., the positive control), which reduced plasma transaminases by ∼43% (p<0.05).
, CCl4 (distilled water+CCl4), LD (low dose of 0.057mgkg−1 liraglutide+CCl4), HD (high dose of 0.118mgkg−1 liraglutide+CCl4), NAC (500mgkg−1 N-acetylcysteine+CCl4). The data are expressed as mean±SEM. The analyses were performed using one-way ANOVA followed by Bonferroni")
Relative weight of the liver in mice that were subjected to CCl4-induced liver injury, expressed as a percentage of body weight. Groups: VEH (distilled water+canola oil), CCl4 (distilled water+CCl4), LD (low dose of 0.057mgkg−1 liraglutide+CCl4), HD (high dose of 0.118mgkg−1 liraglutide+CCl4), NAC (500mgkg−1 N-acetylcysteine+CCl4). The data are expressed as mean±SEM. The analyses were performed using one-way ANOVA followed by Bonferroni's post hoc test. #p<0.05, compared with vehicle group; *p<0.05, compared with CCl4 group.
 ALT with pretreatment protocol. (B) AST with pretreatment protocol. (C) ALP with pretreatment protocol. (D) Glucose with pretreatment protocol. (E) ALT with late treatment protocol. (F) AST with late treatment protocol. (G) ALP with late treatment protocol. (H) Glucose with late pretreatment protocol. Groups: VEH (distilled water+canola oil), CCl4 (distilled water+CCl4), LD (low dose of 0.057mgkg−1 liraglutide+CCl4), HD (high dose of 0.118mgkg−1 liraglutide+CCl4), NAC (500mgkg−1 N-acetylcysteine+CCl4). The data are expressed as mean±SEM. The analyses were performed using one-way ANOVA followed by Bonferroni")
Biochemical parameters in mice that were subjected to CCl4-induced acute liver injury and treated with water, liraglutide, or N-acetylcysteine. (A) ALT with pretreatment protocol. (B) AST with pretreatment protocol. (C) ALP with pretreatment protocol. (D) Glucose with pretreatment protocol. (E) ALT with late treatment protocol. (F) AST with late treatment protocol. (G) ALP with late treatment protocol. (H) Glucose with late pretreatment protocol. Groups: VEH (distilled water+canola oil), CCl4 (distilled water+CCl4), LD (low dose of 0.057mgkg−1 liraglutide+CCl4), HD (high dose of 0.118mgkg−1 liraglutide+CCl4), NAC (500mgkg−1 N-acetylcysteine+CCl4). The data are expressed as mean±SEM. The analyses were performed using one-way ANOVA followed by Bonferroni's post hoc test. #p<0.05, compared with vehicle group; *p<0.05, compared with CCl4 group.
CCl4 administration reduced blood glucose levels by 44% (p=0.010) and 47% (p=0.002) with the pretreatment and late treatment protocols, respectively. The groups that received pretreatment with liraglutide but not late treatment exhibited a tendency to maintain normal blood glucose levels, similar to N-acetylcysteine treatment (Fig. 3C, F). No group differences in hemograms were observed (Supplementary Table S2).
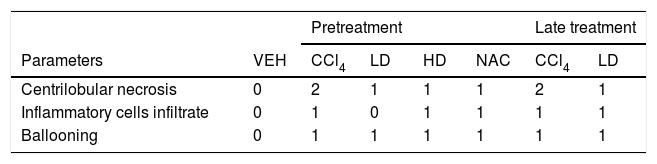
3.2Liraglutide attenuated hepatic histopathological lesionsThe analysis of hepatic tissue showed that CCl4 induced necrosis to a significant degree around the central vein (Fig. 4B, F). A mild degree of hepatocyte ballooning was detected at the interface between healthy and injured tissue. Mild lymphoplasmacytic infiltrates were also observed. Animals that were treated with liraglutide with both protocols exhibited a lower degree of necrosis that was classified as mild (Fig. 4C, D, G). The degree of hepatocyte ballooning and lymphoplasmacytic infiltrates were moderate in all of the groups that received CCl4 (Table 1).
 from representative mice from the following groups: (A) VEH (distilled water+canola oil), (B) CCl4 pretreatment (distilled water+CCl4), (C) LD pretreatment (low dose of 0.057mgkg−1 liraglutide+CCl4), (D) HD pretreatment (high dose of 0.118mgkg−1 liraglutide+CCl4), (E) NAC pretreatment (500mgkg−1 N-acetylcysteine+CCl4), (F) CCl4 late treatment (distilled water+CCl4), (G) LD late treatment (low dose of 0.057mgkg−1 liraglutide+CCl4). *, centrilobular necrosis; →, ballooning hepatocytes. Scale bar=100μm.")
Histological images of liver sections (HE staining) from representative mice from the following groups: (A) VEH (distilled water+canola oil), (B) CCl4 pretreatment (distilled water+CCl4), (C) LD pretreatment (low dose of 0.057mgkg−1 liraglutide+CCl4), (D) HD pretreatment (high dose of 0.118mgkg−1 liraglutide+CCl4), (E) NAC pretreatment (500mgkg−1 N-acetylcysteine+CCl4), (F) CCl4 late treatment (distilled water+CCl4), (G) LD late treatment (low dose of 0.057mgkg−1 liraglutide+CCl4). *, centrilobular necrosis; →, ballooning hepatocytes. Scale bar=100μm.
Histological parameters observed in CCl4-induced liver injury.
| Pretreatment | Late treatment | ||||||
|---|---|---|---|---|---|---|---|
| Parameters | VEH | CCl4 | LD | HD | NAC | CCl4 | LD |
| Centrilobular necrosis | 0 | 2 | 1 | 1 | 1 | 2 | 1 |
| Inflammatory cells infiltrate | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| Ballooning | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
Groups: VEH (distilled water+canola oil), CCl4 (distilled water+CCl4), LD (low dose of 0.057mg/kg liraglutide+CCl4), HD (high dose of 0.118mg/kg liraglutide+CCl4), NAC (500mg/kg N-acetylcysteine+CCl4). Grade of parameters was adapted from Knodel et al.12: Centrilobular necrosis (0 – none; 1 – ≤1/3; 2 – 1/3 to 2/3; 3 – ≥ 2/3), Inflammatory infiltrate and Ballooning (0 – none; 1 – mild; 2 – moderate; 3 – marked).
Hepatic redox homeostasis was altered by CCl4, reflected by reductions of GSH levels and SOD activity (Fig. 5). Liraglutide treatment before and after CCl4-induced hepatic injury preserved GSH levels (Fig. 5A, D) and SOD activity (Fig. 5B, E) at levels that were similar to normal (vehicle-treated) and N-acetylcysteine-treated animals. No changes in GST or CAT activity were observed (data not shown). After CCl4 administration, LPO levels increased by 60% (p=0.002) and 81% (p=0.003) with the liraglutide pretreatment and late treatment protocols, respectively. Pretreatment with liraglutide decreased LPO levels by 28% (∼8.8nmolmin−1mg of protein−1 in liraglutide groups compared to 12.3nmolmin−1mg of protein−1 in CCl4 group) (Fig. 5C), similar to N-acetylcysteine treatment. Late treatment with liraglutide partially reduced LPO levels compared with the CCl4 group (Fig. 5F).
 Glutathione levels with pretreatment. (B) Superoxide dismutase with pretreatment. (C) Lipoperoxidation levels with pretreatment. (D) Glutathione levels with late treatment. (E) Superoxide dismutase with late treatment. (F) Lipoperoxidation levels with late treatment. Groups: VEH (distilled water+canola oil), CCl4 (distilled water+CCl4), LD (low dose of 0.057mgkg−1 liraglutide+CCl4), HD (high dose of 0.118mgkg−1 liraglutide+CCl4), NAC (500mgkg−1 N-acetylcysteine+CCl4). The data are expressed as mean±SEM. The analyses were performed using one-way ANOVA followed by Bonferroni")
Effects of liraglutide on hepatic oxidative stress in CCl4-induced hepatic injury. (A) Glutathione levels with pretreatment. (B) Superoxide dismutase with pretreatment. (C) Lipoperoxidation levels with pretreatment. (D) Glutathione levels with late treatment. (E) Superoxide dismutase with late treatment. (F) Lipoperoxidation levels with late treatment. Groups: VEH (distilled water+canola oil), CCl4 (distilled water+CCl4), LD (low dose of 0.057mgkg−1 liraglutide+CCl4), HD (high dose of 0.118mgkg−1 liraglutide+CCl4), NAC (500mgkg−1 N-acetylcysteine+CCl4). The data are expressed as mean±SEM. The analyses were performed using one-way ANOVA followed by Bonferroni's post hoc test. #p<0.05, compared with vehicle group; *p<0.05, compared with CCl4 group.
All of the liraglutide concentrations had similar and moderate free radical scavenging activity (Fig. 6), although the degree of the antioxidant activity of liraglutide was not as intense as ascorbic acid. The highest liraglutide concentration (1000μgml−1) reduced DPPH radicals by 43% (p=0.0018); the other concentrations reduced DPPH radicals by ∼23% (p=0.0001).
 were used as negative and positive controls, respectively. The data are expressed as the mean±SEM of a triplicate assay. The analyses were performed using one-way ANOVA followed by Bonferroni")
Free radical scavenging activity of liraglutide in the DPPH assay. Distilled water and ascorbic acid (AA, 50μgml−1) were used as negative and positive controls, respectively. The data are expressed as the mean±SEM of a triplicate assay. The analyses were performed using one-way ANOVA followed by Bonferroni's test. #p<0.05, compared with negative control; *p<0.05, compared with positive group.
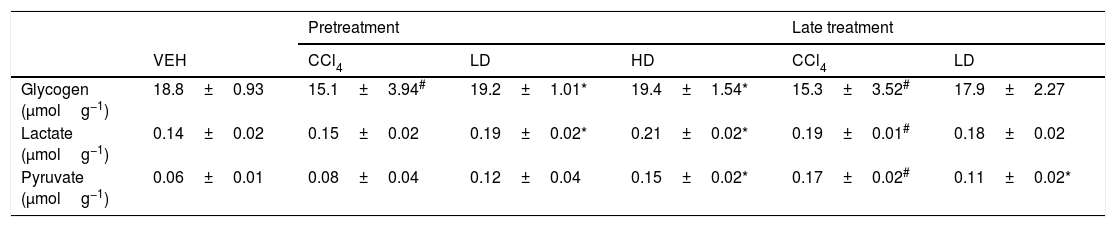
The groups that were challenged with CCl4 exhibited a significant reduction of glycogen levels (∼15.0mmolg of tissue−1). Pretreatment with liraglutide maintained hepatic glycogen (∼19.3mmolg of tissue−1) at levels that were similar to normal control animals (18.8mmolg of tissue−1) (Table 2). However, no effect was observed with the liraglutide late treatment protocol. CCl4-induced hepatic injury only caused significant increases in hepatic lactate and pyruvate concentrations with the late treatment protocol compared with vehicle-treated animals. Pretreatment with liraglutide increased the levels of these metabolites even further, especially at the HD, in which lactate increased by 36% (p=0.018) and pyruvate increased by 77% (p=0.006). The opposite was observed with the late treatment protocol, in which a 37% decrease in pyruvate levels was observed (Table 2).
Hepatic metabolites evaluation after CCl4-induced liver injury.
| Pretreatment | Late treatment | |||||
|---|---|---|---|---|---|---|
| VEH | CCl4 | LD | HD | CCl4 | LD | |
| Glycogen (μmolg−1) | 18.8±0.93 | 15.1±3.94# | 19.2±1.01* | 19.4±1.54* | 15.3±3.52# | 17.9±2.27 |
| Lactate (μmolg−1) | 0.14±0.02 | 0.15±0.02 | 0.19±0.02* | 0.21±0.02* | 0.19±0.01# | 0.18±0.02 |
| Pyruvate (μmolg−1) | 0.06±0.01 | 0.08±0.04 | 0.12±0.04 | 0.15±0.02* | 0.17±0.02# | 0.11±0.02* |
Groups: VEH (distilled water+canola oil), CCl4 (distilled water+CCl4), LD (low dose of 0.057mg/kg liraglutide+CCl4), HD (high dose of 0.118mg/kg liraglutide+CCl4), NAC (500mg/kg N-acetylcysteine+CCl4). The data are expressed as mean±SEM. The analyses were performed using one-way ANOVA followed by Bonferroni's test. #p<0.05, compared with vehicle group; *p<0.05, compared with CCl4 group. Glycogen, lactate and pyruvate were expressed in μmolg of tissue−1.
The gravimetric analysis showed the significant accumulation of hepatic lipids in the CCl4 group compared with the vehicle group with both treatment protocols (Fig. 7A, D). Pretreatment with liraglutide at the LD reduced lipid accumulation (Fig. 7A). Late treatment with liraglutide reduced fat accumulation by 36%, but this reduction was not significant compared with the CCl4 group (Fig. 7D). Both hepatic triglycerides (Fig. 7B, E) and total cholesterol (data not shown) increased in the CCl4 group. Liraglutide treatment reduced the concentrations of these lipids, but the reductions were not statistically significant. A tendency toward a reduction of cholesterol excretion in bile was observed in all of the groups that received CCl4, but no group differences were observed (Fig. 7C, D).
 Hepatic gravimetric analysis with pretreatment. (B) Hepatic triglyceride analysis with pretreatment. (C) Biliary total cholesterol analysis with pretreatment. (D) Hepatic gravimetric analysis with late treatment. (E) Hepatic triglyceride analysis with late treatment. (F) Biliary total cholesterol analysis with late treatment. Groups: VEH (distilled water+canola oil), CCl4 (distilled water+CCl4), LD (low dose of 0.057mgkg−1 liraglutide+CCl4). The data are expressed as mean±SEM. The analyses were performed using one-way ANOVA followed by Bonferroni")
Effects of liraglutide on hepatic and biliary lipids. (A) Hepatic gravimetric analysis with pretreatment. (B) Hepatic triglyceride analysis with pretreatment. (C) Biliary total cholesterol analysis with pretreatment. (D) Hepatic gravimetric analysis with late treatment. (E) Hepatic triglyceride analysis with late treatment. (F) Biliary total cholesterol analysis with late treatment. Groups: VEH (distilled water+canola oil), CCl4 (distilled water+CCl4), LD (low dose of 0.057mgkg−1 liraglutide+CCl4). The data are expressed as mean±SEM. The analyses were performed using one-way ANOVA followed by Bonferroni's test. #p<0.05, compared with vehicle group; *p<0.05, compared with CCl4 group.
The present results showed that liraglutide exerted protective and therapeutic actions against acute hepatotoxicity. Liraglutide decreased plasma ALT, AST, and ALP levels and liver necrosis when administered both before and after CCl4-induced liver injury. These results indicate a hepatic action of liraglutide and corroborate previously published studies that used models of nonalcoholic steatohepatitis and ischemia–reperfusion injury [7,23,24]. A previous study showed that exenatide, the GLP-1 analog precursor, exerted beneficial actions against hepatic oxidative stress and steatosis in metabolic disease [25]. Thus, the present study investigated the possible mechanisms of the protective and therapeutic effects of liraglutide against CCl4-induced hepatic injury.
Trichloromethyl radicals that are overproduced by the hepatic metabolism of CCl4 were shown to negatively influence cellular antioxidant balance, promoting lipid peroxidation, causing GSH depletion, and reducing antioxidant enzyme activity [26,27]. GLP-1 analogs, including liraglutide, have been reported to be possible synthetic therapeutic alternatives to reduce oxidative stress and consequently attenuate cell death in gastric [28], neuronal [29], renal [30], and hepatic cells in chronic disorders [7,8,23]. The present study found that liraglutide decreased LPO levels and increased GSH levels and SOD activity in a mouse model of acute hepatic injury. GSH is important for protecting cells against toxicity that is caused by peroxides and other free radicals [31]. Superoxide dismutase is an antioxidant enzyme that interacts with superoxide radicals (O2−) and promotes their neutralization through H2O2 formation, which is subsequently eliminated by other enzymes [32]. The hepatic effects of liraglutide in the present study are similar to hepatoprotective effects of N-acetylcysteine, which was shown to maintain redox balance by modulating GSH [33]. The per se antioxidant potential of liraglutide was also demonstrated (Fig. 6). Overall, these data suggest that liraglutide modulates the hepatic redox system through two mechanisms: 1 – preserving the activity of hepatic antioxidant components and 2 – neutralizing free radicals that are generated during the disease process.
The liver is an integral organ for metabolism. Several metabolites were analyzed in the present study to evaluate the effects of liraglutide. Toxicity that was induced by CCl4 depleted plasma glucose levels. Liraglutide treatment with both protocols maintained glucose levels similar to vehicle-treated animals (Fig. 3C, F). The decrease in plasma glucose levels could indicate a greater liver energy demand [34]. A significant decrease in hepatic glycogen levels also occurred after CCl4 exposure (Table 2), suggesting intense rates of glycolysis and glycogenolysis that compensate for blood hypoglycemia, which has been associated with hepatic failure [35,36]. Higher levels of lactate and pyruvate in liver homogenates that resulted from glycolysis were also observed after CCl4 administration (Table 2). Similar hepatic metabolism results were reported in a previous study that evaluated acetaminophen-induced lesion [35]. Liraglutide, despite being a hypoglycemic drug, exerted a protective effect and maintained blood glucose levels similar to control animals. This finding is related to its glucose state-dependent action, in which it exerts effects only when glycemia levels are high. This supports the possibility of using liraglutide for other therapeutic purposes without affecting glycemia [37]. However, this normoglycemic effect was not observed when liraglutide was administered after hepatic injury. Animals that were pretreated with liraglutide maintained hepatic glycogen at levels that were similar to vehicle-treated animals. Pretreatment with liraglutide preserved hepatic glycometabolism, which was also reported in a previous study in diabetic mice [38]. Valverde et al. (1994) performed an in vitro assay and found that incretins, including GLP-1, increased glycogen synthesis [39].
Pretreatment with liraglutide also suppressed hepatic lipid accumulation that was caused by CCl4[40,41]. Mice with nonalcoholic fatty liver disease that were treated with liraglutide exhibited an improvement in hepatic lipid congestion [42], as well as the benefits of liraglutide in human patients with type 2 diabetes who developed steatosis [43], thus supporting our findings. However, these interesting gravimetric results were not reproduced in the differentiated lipid analysis in liver tissue, in which only discrete changes in hepatic triglycerides and total cholesterol were observed. Liraglutide administration has been previously reported to be a therapeutic alternative to reverse high triglyceride levels in nonalcoholic fatty liver disease [44,45]. Liraglutide-treated animals in the present study exhibited lower levels of triglycerides, but this decrease was not statistically significant. Other lipids that are present in the liver are likely altered by CCl4 and liraglutide. Further studies of various lipids should be performed to clarify this issue. Interestingly, liraglutide does not appear to interfere with the bile excretion of cholesterol. Liraglutide may reduce steatosis by improving lipolysis or attenuating lipogenesis rather than by increasing lipid excretion from the liver to bile.
In conclusion, the present study found hepatoprotective and therapeutic effects of liraglutide against acute CCl4-induced hepatic injury. The mechanism of action of liraglutide appears to be related to the modulation of oxidative stress and hepatic metabolism (Fig. 8). These effects appear to be independent of dose, in which both doses that were tested in the present study exerted similar effects. Overall, liraglutide may have potential as a complementary therapy for the treatment of drug-induced liver injury.AbbreviationsCCl4
carbon tetrachloride
i.p.intraperitoneal
CCl3trichloromethyl radical
OOCl3trichloromethyl peroxide radical
GLP-1glucagon-like peptide-1
Nrf-2nuclear factor erythroid 2-related factor 2
LDlow dose
HDhigh dose
ASTaspartate transaminase
ALTalanine transaminase
ALPalkaline phosphatase
HEhematoxylin and eosin
GSHreduced glutathione
LPOlipoperoxidation
GSTglutathione S-transferase
SODSuperoxide dismutase
CATcatalase
DPPH2,2-diphenyl-1-picrylhydrazyl
NACN-acetylcysteine
NAD+oxidized nicotinamide adenine dinucleotide
NADHreduced oxidized nicotinamide adenine dinucleotide
ANOVAone-way analysis of variance
O2−superoxide radical
H2O2hydrogen peroxide
NACN-acetylcysteine
VEHvehicle
AAascorbic acid

Liraglutide hepatic effects against CCl4 acute injury. Legend – CCl4: carbon tetrachloride; CCl3: trichoromethyl radical; CYP: cytochrome P; O2: molecular oxygen; CCl3OO: trichoromethyl peroxide radical; LPO: lipoperoxidation; O2−: superoxide anion; H2O2: hydrogen peroxide; H2O: water; OH: hydroxyl anion; Fe: iron; SOD: superoxide dismutase; CAT: catalase; GSH: reduced glutathione; GPX: glutathione peroxidase; GR: glutationa reductase; GSSH: oxidized glutathione.
This study was financed by (CAPES) Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Finance Code 001) and (CNPq)Conselho Nacional de Desenvolvimento Científico e Tecnológico (no. 037977/2015-3).
Conflict of InterestedThe authors declare no conflict of interest.
The authors thank Rafaela Santa Clara, Liziane Malaquias, and Gabriele Seko for assistance with the experiments and Prof. Jurandir Comar for providing enzymes. The authors thank CTAF-UFPR for the histological images.
The following are the supplementary data to this article:



