



Hepatobiliary transport systems mediate hepatic uptake and biliary excretion of bile acids, bilirubin and other biliary constituents. Hereditary or acquired defects of these transporters may cause or maintain cholestasis and jaundice under various clinical conditions including progressive familial intrahepatic cholestasis (PFIC) 1-3 or its milder forms, benign recurrent intrahepatic cholestasis (BRIC) 1 and 2, Dubin-Johnson syndrome, drug and inflammation-induced cholestasis and intrahepatic cholestasis of pregnancy. Moreover, induction of alternative efflux pumps for bile acids/bilirubin and phase I/II detoxifying enzymes may counteract hepatic accumulation of potentially toxic biliary constituents in cholestasis by providing alternative escape routes. Transcriptional and post-transcriptional regulation of hepatobiliary transporters in health and disease is mediated by multiple factors such as bile acids, proinflammatory cytokines, drugs and hormones. Ligand-activated nuclear receptors (NR) and hepatocyte-enriched transcription factors play a critical role in transcriptional transporter regulation. Many hepatobiliary transporter alterations in cholestatic liver disease can now be explained by ligand binding of accumulating cholephiles to NRs. Moreover, NRmediated actions may be targeted by pharmacological ligands. Understanding the transcriptional mechanisms leading to transporter changes therefore not only represents a key for understanding the pathophysiology of the cholestatic liver disease, but also represents a prerequisite for designing novel therapeutic strategies.
Abbreviations:
ABC, ATP-binding cassette
CAR (NR1I3), constitutive androstane receptor
CBDL, common bile duct ligation
Cyp, cytochrome p450
FXR (NR1H4), farnesoid X receptor/bile acid receptor
HNF, Hepatocyte nuclear factor
Mdr, multidrug resistance gene
Mrp, multidrug resistance-associated protein
Ntcp (Slc10a1), Na+/ taurocholate cotransporter
Oatp (Slc21a), organic anion transporter
PPAR á (NR1C1), peroxisome proliferator activated receptor alpha
PXR (NR1I2), pregnane X receptor
RARá (NR1B1), retinoic acid receptor alpha
RXRá (NR2B1), retinoid X receptor alpha
SHP (NR0B2), short heterodimer partner
Financial support: This work was supported by grant P15502 from the Austrian Science Fund, grant 10266 from the Jubilee Funds of the Austrian National Bank and a GEN-AU grant from the Austrian Ministry for Science (to M.T.).
IntroductionSince the cloning of the major hepatic bile acid uptake system as the first hepatobiliary bile acid transport system in 1991,1 a substantial amount of further hepatocellular membrane proteins involved in organic anion and bile acid transport together with their substrates have been characterized.2-4 Ever since numerous hereditary as well as acquired cholestatic liver diseases have been linked to mutated or modified transporter expression.2,5 A complex regulatory network at a transcriptional, posttranscriptional, translational and post-translational level mediates the phys-iological and pathophysiological stimuly resulting in causative or adaptive transporter changes in liver disease.6-11 While the understanding of the molecular and biochemical mechanisms leading to human liver diseases is becoming more and more detailed an effective treatment is still elusive. This review therefore focuses on recent aspects of transcriptional hepatobiliary transporter regulation under physiological and pathophysiological conditions with a special emphasis on regulation by nuclear transcription factors and their implications for future “customized” treatment strategies in cholestatic disorders.
The Enterohepatic circulationPrinciples of bile formationGeneration of bile flow is an active process driven by the excretion of organic solutes (bile acids, cholesterol and phospholipids) into the canalicular space between adjacent hepatocytes followed by passiv para-and transcellular flux of filterable solutes (e.g. electrolytes, glucose, amino acids) and water. Conjungated bile acids, the major biliary organic solutes are highly concentrated up to 1,000 fold in bile via active transport mechanisms and their vectorial transport from sinusoidal blood to bile represents the major driving force for the so-called bile salt dependent bile flow. Therefore, concentrations of bile acids in the canaliculus and ductules are high (about 20-50 mmol/L) and as high as 300 mmol/L in the gallbladder, while they are diluted to 1-10 mmol/L in the gastrointestinal tract and are as low as 20 - 50,amol/L in the portal vein and only 5,amol/L after efficient first pass extraction in the systemic venous plasma.12 Biliary bile acids then induce canalicular phospholipid and cholesterol secretion and form mixed biliary micelles. Bile salt independent bile flow consists mainly of the canalicular secretion of reduced glutathione and the excretion of bicarbonate. The canalicular primary bile is further modified by absorptive and secretory processes along the biliary tree. Considerable species specific differences in bile formation exist including the contribution of ductular bile and bile acid composition.2,13-15
Bile acids undergo an enterohepatic circulation which consists of their continuous hepatocellular canalicular se-cretion, active re-absorption in the terminal ileum and to lower degrees passive absorption in the colon, and hepatic basolateral re-uptake. Thus, bile acids are getting physiologically accumulated and this bile salt pool in human averages 3-4g and circulates 6-10 times a day. Apart from this major route for bile acids, also cholehepatic shunting (i.e. recirculation of bile acids from the bile duct lumen via cholangiocytes and the peribiliary plexus) and renal-hepatic circulation (i.e. glomerular filtration of bile acids from plasma to urine and reabsorption in the proximal tubule to minimize renal bile acid loss) of bile components occurs, processes which, however gain their full importance mainly under pathological cholestatic conditions.2,12 Because of the efficient cycling through liver and intestine as well as minimzing renal loss only 0.5g of bile acids are lost daily by fecal excretion and have to be restored by endogenous conversion of cholesterol to bile acids.
The physilogical role of bile comprises digestion and absorption of intestinal lipids and excretion of endogenous (e.g. cholesterol, bile acids, bilirubin) as well as exogenous (e.g. environmental toxins, drugs and their metabolites) compounds. For a long time the centerpiece in “bile-ology” was thought to be the regulation of cholesterol homeostasis, which is mediated either by biliary secretion of free cholesterol (two thirds) or after conversion of cholesterol into bile acids (one third). However, more and more it turns out that bile acids are not only the waste product of cholesterol, which in turn stimulates bile flow and serves as detergent for dietary fats and vitamines, but have a broad spectrum as signaling molecules with hormone like attitutes. As such bile acids ligand-activate several nuclear receptors involved in bile formation and secretion, detoxification reactions of bile acids and xenobiotics and are even involved in apoptosis or the glucose metabolism.2,3,16,17 Thus, it is not surprising that the key step in the formation of bile acid from cholesterol is regulated by complex feedforward and feedback mechanisms.11,18
Basically, bile acids are formed of cholesterol exclusively in pericentral hepatocytes by either the neutral (classic) pathway resulting in the formation of the primary bile acids cholic acid (CA) and chenodeoxycholic acid (CDCA) in roughly equal amounts or the acidic (alternative) pathway, which mainly produces CDCA and may become a more important route in chronic liver disease.18-21 Before bile acids are excreted they are conjungated with either glycine or taurine. In the distal intestine the primary bile acids CA and CDCA are deconjungated and deoxygenated by anerobic bacteria to form the secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA). To some extents DCA and LCA are absorbed in the colon and re-conjugated in the hepatocytes. While DCA can constitute up to 20% of total biliary bile acids in man, LCA is additionally sulfated, which limits its intestinal reabsorption. Thus, the highly hepatotoxic LCA constitutes no more than 5% of biliary bile acids.12 In humans also trace amounts of the 7/3-epimer of CDCA, the therapeutically used ursodeoxycholic acid (UDCA) is formed. In cholestatic liver diseases accumulation of bile acids is counteracted to a certain degree by additional hydroxylation, glucuronidation or sulfation reactions, providing a more hydrophilic water soluble bile acid pool, which in turn can be more efficiently excreted via urine.22-27
Hepatobiliary bile acid transportBasolateral bile acid uptake and alternative export: Bile acids return to the basolateral (sinusoidal) membrane of hepatocytes mostly in albumin bound form. While dihydroxy bile acids are almost completely albumin bound, protein binding decreases with degree of hydroxilation.12 These bile acid-albumin complexes pass the fenestrae of the sinusoidel epithelium into the space of Disse, where albumin is dissociated either by the microenvironment of the Disse space or the sinusoidel membrane itself.28-31 Hepatic first pass effect of conjugated bile acids ranges from 75-90% depending on their structure.28 Under normal conditions this uptake occurs in periportal hepatocytes of zone 1, under cholestatic conditions with higher bile acid concentrations also hepatocytes in zone 3 are recruited.32 Unconjungated patocyte predominatly by passive diffusion or via Na+-independent mechanisms, conjungated bile acids largely by Na+-dependent mechanisms. The Na+ taurocholate co-transporting polypeptide (NTCP) accounts for more than 80% uptake of taurocholate but less than 50% of unconjugated cholate, thus taurocholate is more than 10fold concentrated in hepatocytes.28 The basolateral Na+/K+- ATPase maintains a transmembranous Na+ gradient which is the driving force for this Na+-dependent transport.4 NTCP is exclusively located to the basolateral membrane of hepatocytes throughout the liver and functionally transports all physiological bile acids and to small extents also other compounds33(Figure 1,Table I). The bulk of Na+-independent bile acid transport is mediated by members of the family of organic anion transporter proteins (OATP/SLC21A - please note the new nomenclature for OATP/Oatp proposed in Hagenbuch et al.),34 which is driven by glutathione and/or bicarbonate anion exchange. In contrast to NTCP, OATPs are not restricted solely to hepatocytes but are also expressed in multiple tissues including kidney, neuronal structures and intestine. In addition, OATPs also have broader substrate preferences including conjugated and unconjugated bile acids, bilirubin, neutral steroids, eicasanoids, numerous xenobiotics and many more.34 In rodents Oatp1/Slc21a1 (new nomenclature Oatp1a1) mediates most of the bile acid transport, while Oatp2/Slc21a4 (new nomenclature Oatp1a4) and Oatp4/Slc21a10 (new nomenclature Oatp1b2) are of minor importance. In contrast to the homogenous distribution of Oatp1, Oatp2 is predominantly localized to the pericentral region, thus being a potential backup when unextracted bile salts spill over from the periportal region under cholestatic conditions. Among human OATPs, which are not true homologs of the rodent counterparts, OATP2/OATP-C/ SLC21A6 (new nomenclature OATP1B1) represents the most important bile salt transporter, while the contribution of OATP-A/SLC21A3 (new nomenclature OATP1A2) is minor and OATP-B/SLC21A9 (new nomenclature OATP2B1) does not transport bile acids. The role of OATP-8/SLC21A8 (new nomenclature OATP1B3), a gene duplication variant of OATP2, in bile acid transport is still controversial.2,4,13 In addition, organic anion transporters (Oat), localized to the basolateral membrane of kidneys and one isoform (Oat2) also to hepatocytes may additionally modulate hepatic drug and bile acid uptake, although specific bile acid transport has so far only been shown for the renal isoforms.35
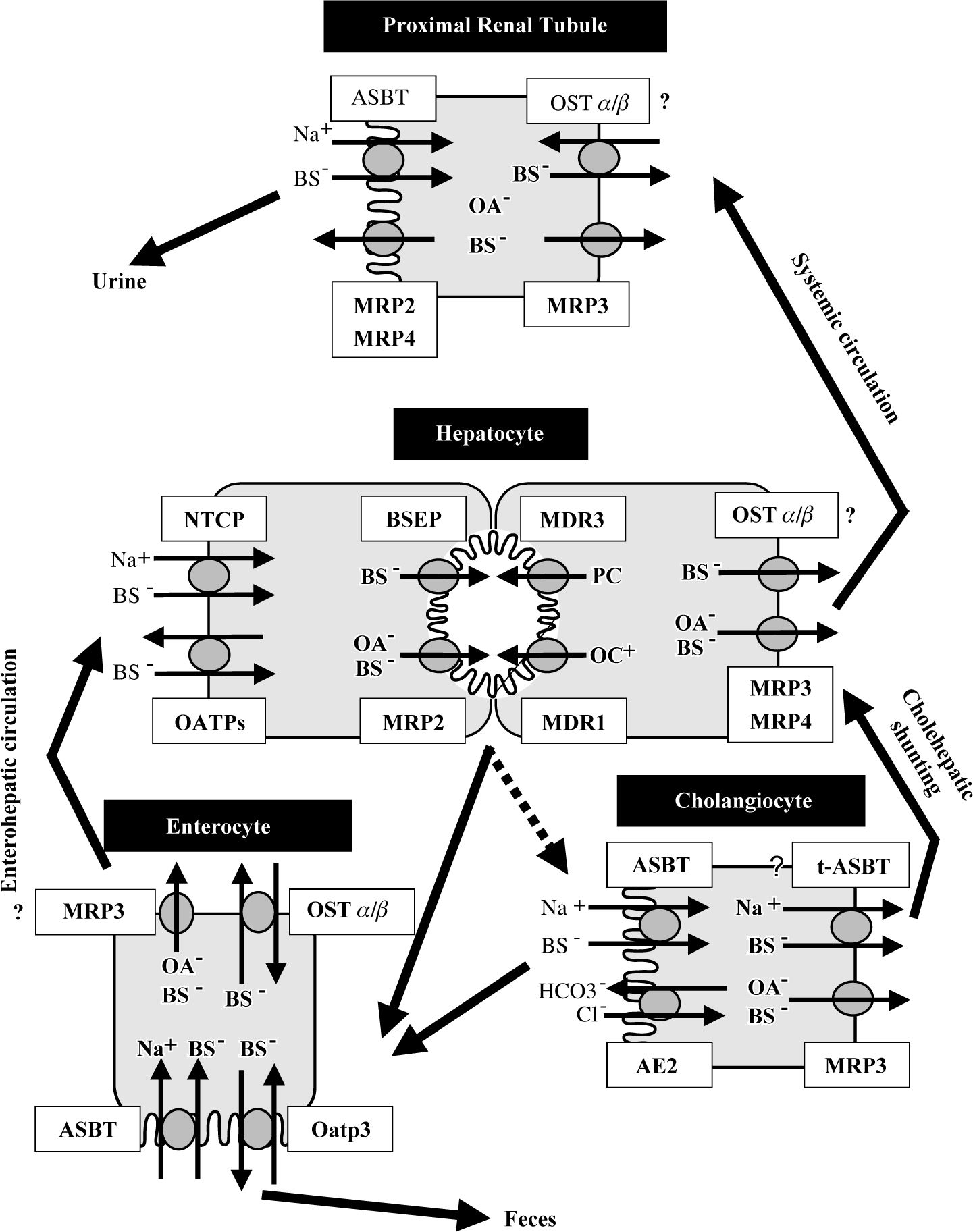
 and Na+-independent (OATPs) transport systems. Monovalent bile acids are excreted into the bile canaliculus via BSEP – determining bile salt dependent bile flow-, while divalent bile acids and anionic conjugates (e.g. bilirubin diglucuronide, glutathione) are excreted via MRP2 – determining bile salt independent bile flow. MDR3 mediates the canalicular secretion of phospholipids, which form mixed micelles together with bile acids and cholesterol. MDR1 excretes the bulky of organic cations. Basolateral bile acid export pumps (e.g. MRP3, MRP4) provide an alternative excretory pathway for otherwise accumulated biliary constituents. Bile composition is further modified along the biliary passage by secretion of bicarbonate via AE2 and reabsorption of bile acids via luminal ASBT. A shortcut (called “cholehepatic shunting”) between cholangiocytes and hepatocytes is proposed by basolateral export of bile acids from the cholangiocytes via MRP3 and possibly t-ASBT and hepatocellular reuptake. In the terminal ilieum bile acids are reabsorbed by ASBT and to certain degrees in rodents by Oatp3 and effluxed on the basolateral pole of enterocytes via OSTα/β and possibly to a lesser extent via MRP3 into portal circulation. Similar to the cholangiocyte and enterocyte, in the proximal renal tubules, bile acids are reabsorbed from the glomerular filtrate via ASBT to minimize bile salt loss. Possibly, renal MRP2 and MRP4 may be involved in the secretion of bile acids into the urine under cholestatic conditions.")
Hepatobiliary transporters in liver and extraheptic tissues. Bile acids are taken up by the hepatocytes via Na+-dependent (NTCP) and Na+-independent (OATPs) transport systems. Monovalent bile acids are excreted into the bile canaliculus via BSEP – determining bile salt dependent bile flow-, while divalent bile acids and anionic conjugates (e.g. bilirubin diglucuronide, glutathione) are excreted via MRP2 – determining bile salt independent bile flow. MDR3 mediates the canalicular secretion of phospholipids, which form mixed micelles together with bile acids and cholesterol. MDR1 excretes the bulky of organic cations. Basolateral bile acid export pumps (e.g. MRP3, MRP4) provide an alternative excretory pathway for otherwise accumulated biliary constituents. Bile composition is further modified along the biliary passage by secretion of bicarbonate via AE2 and reabsorption of bile acids via luminal ASBT. A shortcut (called “cholehepatic shunting”) between cholangiocytes and hepatocytes is proposed by basolateral export of bile acids from the cholangiocytes via MRP3 and possibly t-ASBT and hepatocellular reuptake. In the terminal ilieum bile acids are reabsorbed by ASBT and to certain degrees in rodents by Oatp3 and effluxed on the basolateral pole of enterocytes via OSTα/β and possibly to a lesser extent via MRP3 into portal circulation. Similar to the cholangiocyte and enterocyte, in the proximal renal tubules, bile acids are reabsorbed from the glomerular filtrate via ASBT to minimize bile salt loss. Possibly, renal MRP2 and MRP4 may be involved in the secretion of bile acids into the urine under cholestatic conditions.
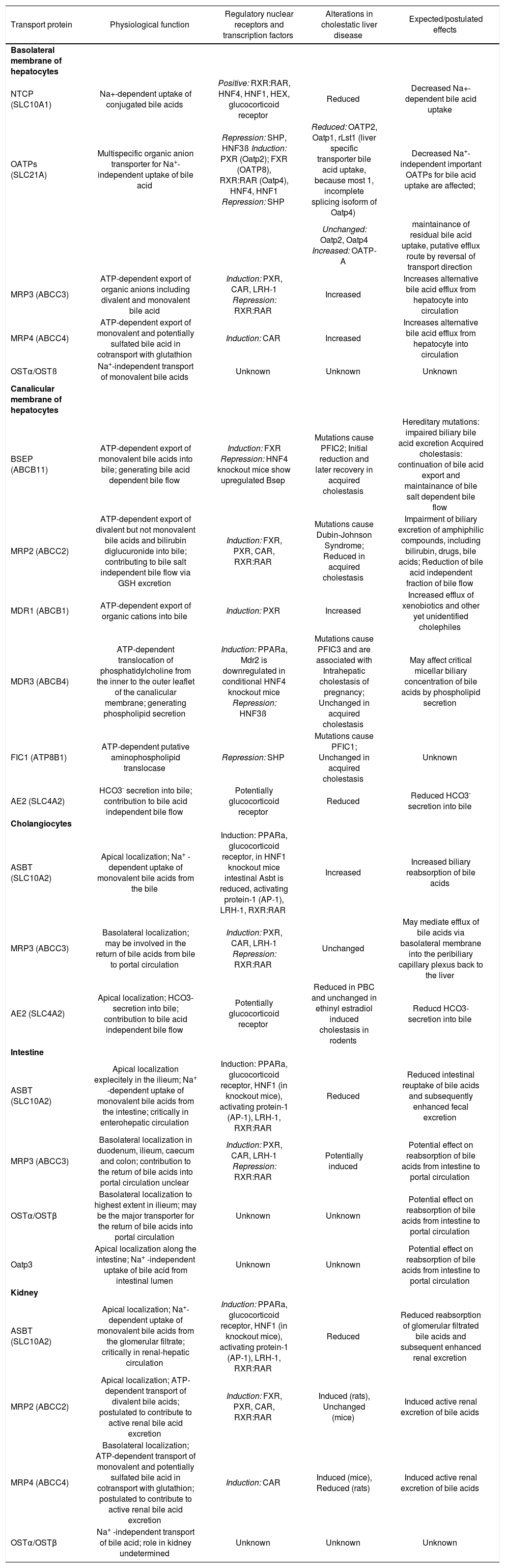
Major hepatobiliary transporters, their function, regulating nuclear receptors, changes in cholestasis and their expected effects in liver and extrahepatic tissue.
| Transport protein | Physiological function | Regulatory nuclear receptors and transcription factors | Alterations in cholestatic liver disease | Expected/postulated effects |
|---|---|---|---|---|
| Basolateral membrane of hepatocytes | ||||
| NTCP (SLC10A1) | Na+-dependent uptake of conjugated bile acids | Positive: RXR:RAR, HNF4, HNF1, HEX, glucocorticoid receptor | Reduced | Decreased Na+-dependent bile acid uptake |
| OATPs (SLC21A) | Multispecific organic anion transporter for Na+-independent uptake of bile acid | Repression: SHP, HNF3ß Induction: PXR (Oatp2); FXR (OATP8), RXR:RAR (Oatp4), HNF4, HNF1 Repression: SHP | Reduced: OATP2, Oatp1, rLst1 (liver specific transporter bile acid uptake, because most 1, incomplete splicing isoform of Oatp4) | Decreased Na+-independent important OATPs for bile acid uptake are affected; |
| Unchanged: Oatp2, Oatp4 Increased: OATP-A | maintainance of residual bile acid uptake, putative efflux route by reversal of transport direction | |||
| MRP3 (ABCC3) | ATP-dependent export of organic anions including divalent and monovalent bile acid | Induction: PXR, CAR, LRH-1 Repression: RXR:RAR | Increased | Increases alternative bile acid efflux from hepatocyte into circulation |
| MRP4 (ABCC4) | ATP-dependent export of monovalent and potentially sulfated bile acid in cotransport with glutathion | Induction: CAR | Increased | Increases alternative bile acid efflux from hepatocyte into circulation |
| OSTα/OSTß | Na+-independent transport of monovalent bile acids | Unknown | Unknown | Unknown |
| Canalicular membrane of hepatocytes | ||||
| BSEP (ABCB11) | ATP-dependent export of monovalent bile acids into bile; generating bile acid dependent bile flow | Induction: FXR Repression: HNF4 knockout mice show upregulated Bsep | Mutations cause PFIC2; Initial reduction and later recovery in acquired cholestasis | Hereditary mutations: impaired biliary bile acid excretion Acquired cholestasis: continuation of bile acid export and maintainance of bile salt dependent bile flow |
| MRP2 (ABCC2) | ATP-dependent export of divalent but not monovalent bile acids and bilirubin diglucuronide into bile; contributing to bile salt independent bile flow via GSH excretion | Induction: FXR, PXR, CAR, RXR:RAR | Mutations cause Dubin-Johnson Syndrome; Reduced in acquired cholestasis | Impairment of biliary excretion of amphiphilic compounds, including bilirubin, drugs, bile acids; Reduction of bile acid independent fraction of bile flow |
| MDR1 (ABCB1) | ATP-dependent export of organic cations into bile | Induction: PXR | Increased | Increased efflux of xenobiotics and other yet unidentified cholephiles |
| MDR3 (ABCB4) | ATP-dependent translocation of phosphatidylcholine from the inner to the outer leaflet of the canalicular membrane; generating phospholipid secretion | Induction: PPARa, Mdr2 is downregulated in conditional HNF4 knockout mice Repression: HNF3ß | Mutations cause PFIC3 and are associated with Intrahepatic cholestasis of pregnancy; Unchanged in acquired cholestasis | May affect critical micellar biliary concentration of bile acids by phospholipid secretion |
| FIC1 (ATP8B1) | ATP-dependent putative aminophospholipid translocase | Repression: SHP | Mutations cause PFIC1; Unchanged in acquired cholestasis | Unknown |
| AE2 (SLC4A2) | HCO3- secretion into bile; contribution to bile acid independent bile flow | Potentially glucocorticoid receptor | Reduced | Reduced HCO3- secretion into bile |
| Cholangiocytes | ||||
| ASBT (SLC10A2) | Apical localization; Na+ -dependent uptake of monovalent bile acids from the bile | Induction: PPARa, glucocorticoid receptor, in HNF1 knockout mice intestinal Asbt is reduced, activating protein-1 (AP-1), LRH-1, RXR:RAR | Increased | Increased biliary reabsorption of bile acids |
| MRP3 (ABCC3) | Basolateral localization; may be involved in the return of bile acids from bile to portal circulation | Induction: PXR, CAR, LRH-1 Repression: RXR:RAR | Unchanged | May mediate efflux of bile acids via basolateral membrane into the peribiliary capillary plexus back to the liver |
| AE2 (SLC4A2) | Apical localization; HCO3-secretion into bile; contribution to bile acid independent bile flow | Potentially glucocorticoid receptor | Reduced in PBC and unchanged in ethinyl estradiol induced cholestasis in rodents | Reducd HCO3-secretion into bile |
| Intestine | ||||
| ASBT (SLC10A2) | Apical localization explecitely in the ilieum; Na+ -dependent uptake of monovalent bile acids from the intestine; critically in enterohepatic circulation | Induction: PPARa, glucocorticoid receptor, HNF1 (in knockout mice), activating protein-1 (AP-1), LRH-1, RXR:RAR | Reduced | Reduced intestinal reuptake of bile acids and subsequently enhanced fecal excretion |
| MRP3 (ABCC3) | Basolateral localization in duodenum, ilieum, caecum and colon; contribution to the return of bile acids into portal circulation unclear | Induction: PXR, CAR, LRH-1 Repression: RXR:RAR | Potentially induced | Potential effect on reabsorption of bile acids from intestine to portal circulation |
| OSTα/OSTβ | Basolateral localization to highest extent in ilieum; may be the major transporter for the return of bile acids into portal circulation | Unknown | Unknown | Potential effect on reabsorption of bile acids from intestine to portal circulation |
| Oatp3 | Apical localization along the intestine; Na+ -independent uptake of bile acid from intestinal lumen | Unknown | Unknown | Potential effect on reabsorption of bile acids from intestine to portal circulation |
| Kidney | ||||
| ASBT (SLC10A2) | Apical localization; Na+- dependent uptake of monovalent bile acids from the glomerular filtrate; critically in renal-hepatic circulation | Induction: PPARa, glucocorticoid receptor, HNF1 (in knockout mice), activating protein-1 (AP-1), LRH-1, RXR:RAR | Reduced | Reduced reabsorption of glomerular filtrated bile acids and subsequent enhanced renal excretion |
| MRP2 (ABCC2) | Apical localization; ATP-dependent transport of divalent bile acids; postulated to contribute to active renal bile acid excretion | Induction: FXR, PXR, CAR, RXR:RAR | Induced (rats), Unchanged (mice) | Induced active renal excretion of bile acids |
| MRP4 (ABCC4) | Basolateral localization; ATP-dependent transport of monovalent and potentially sulfated bile acid in cotransport with glutathion; postulated to contribute to active renal bile acid excretion | Induction: CAR | Induced (mice), Reduced (rats) | Induced active renal excretion of bile acids |
| OSTα/OSTβ | Na+ -independent transport of bile acid; role in kidney undetermined | Unknown | Unknown | Unknown |
Retrograde hepatocellular bile acid efflux via the basolateral membrane may become an important alternative spill over route for accumulating bile acids/bilirubin during cholestatic disorders. The most important adaptive transport systems ruled out so far belong to the class of multidrug resistance ATP-binding cassette (ABC) transporters (MRPs). Mrp1/Abcc1/MRP1/ABCC1, Mrp3/ Abcc3/MRP3/ABCC3 and Mrp2/Abcc4/MRP4/ABCC4 are constitutively expressed only at very low levels at the basolateral hepatocyte membrane, but are inducible under cholestatic conditions. Their substrate specifity comprises divalent bile acids and in the case of MRP3 and MRP4 also monovalent bile acids.2,35-38 Also MRP5/ABCC5 and MRP6/ABCC6 are located to the basolateral membrane of hepatocytes, however, their role in adaptive bile salt transport remains to be further explored.
Under bile acid burden also members of the Oatp family remain candidates for supporting bile acid efflux, since Oatp1 and Oatp2 were shown to act as bidirectional exchangers in Xenopus laevis.39 The new kid in bile acid transport is the heteromeric organic solute transporter OSTa and OSTj3 which was recently shown to transport taurocholate in a Na+-independent manner. However, only the human isoforms were shown to be expressed in hepatocytes as well.40 The murine isoforms were abundantly expressed in small intestine and to lesser extents in kidney, where their major function is adressed to be the basolateral counterpart of apical Asbt.41
Canalicular bile acid excretion: Once being taken up at the basolateral membrane, bile acids are transferred to the canalicular membrane for rapid excretion.4 Under physiological conditions hepatocellular bile acid binding proteins, so far 3a-hydroxysteroid dehydrogenase (3a-HSD), glutathione S transferases (Gst) and the liver fatty acid binding proteins (L-FABP), are involved in this intracellular transfer.42 Under conditions of high bile acid load hydrophobic bile acids may also be transferred within membrane-bound compartements.2,4 In bile formation canalicular bile acid excretion represents the rate limiting step. Similar to the basolateral membrane the most important export pumps for canalicular bile acid excretion belong to the class of ATP-dependent ABC transporter systems. Under normal conditions the bile salt export pump, Bsep/Abcb11/BSEP/ ABCB11, represents the major bile acid transport systems for monovalent bile acids, actively transporting its substrates against a 1,000-fold concentration gradient. While the loss of BSEP in humans, causing hereditary progressive familial intrahepatic cholestasis type 2 (PFIC2), results in a decrease of biliary bile acid concentration to 1% of the normal range,43 the knock-out of Bsep/Abcb11 in mice only results in a decrease of 30% percent44 suggesting the presence of other canalicular bile acid transport systems. Although these additional transport systems have not been identified so far, Mrp2/Abcc2 is a candidate for transporting divalent, glucuronidated and sulfated45 and also tetrahydroxilated bile acids.2 In addition, bilirubin, GSH and a broad range of amphipatic compounds are excreted by Mrp2 as well, however, monvalent bile acids are no substrates for Mrp2.46 An additional transport systems for sulfated conjugates, including dehydro-epiandrosteronsulfate and estrone-3 sulfate is the breast cancer related protein (Bcrp/Abcg2).47 After secretion biliary bile acids drive the secretion of phosphatidylcholine and cholesterol from the outer leaflet of the canalicular membrane. The continous support of phospholipids from the inner to the outer leaflet is mediated by the phospholipid flippase Mdr2/Abcb4 or its human ortholog MDR3/ABCB4.48 The canalicular transport of cholesterol is most likely mediated by the sterol half-transporters Abcg5/8.49 Together bile acids, phospholipids and cholesterol form mixed micelles which in turn avoid bile acid toxicity to the bile duct epithelium.2 Additional canalicular transporters include the multidrug export pump MDR1/ABCB1 (in rodentsMdr1a/Mdr1b) which accounts for the transport of bulky amphipathic cations (e.g. drugs) and the putative aminophospholipid transporter FIC-1/ATP8B1, which mutations causes familial intrahepatic cholestasis type 1.2 The most relevant ATP-independent transporter located to the canalicular membrane is a Cl-/HCO3-exchanger (AE2/SLC4A2) which together with MRP2 accounts for the greatest part of the canalicular bile acid independent bile fraction.50
Cholangiocellular bile acid transportCholangiocytes are important modulators of canalicular bile by secreting and reabsorbing processes while bile passes the intrahepatic biliary tree. Experiments in rats suggest that only large bile ducts (>15,am diameter) contribute to these changes while small bile ducts remain passive.51 While the small amount of monomeric unconjugated bile acids may passively enter cholangiocytes in exchange for HCO3-, thus inducing bicarbonate rich hypercholeresis, a mechanism called cholehepatic shunting and inducible by dihydroxy bile acids and C-23 Nor-dihydroxy bile acids,4,52 the majority of biliary bile acids is conjugated and can be reabsorbed in a Na+-dependent manner via the apical bile acid transporter Asbt.53 After uptake and potential conjugation during transcellular transport bile acids may be effluxed to the plasma of the periductular capillary plexus via an anion exchange mechanism.54 Candiates for this mechanism are the human OATP-A55 or the rodent Oatp3/Slc21a7 (new nomenclature Oatp1a5).13 In addition, this could also be achieved by Mrp3/MRP3, which is located to the basolateral membrane of cholangiocytes 56,57 or a splicing variant of Asbt/SLC10A2 so called truncated Asbt (t-Asbt).58 Similar to the function of Osta/Ostfi in the intestine41 it could well be imagined that this heterodimeric transport systems functions also in the cholangiocytes as counterpart of Asbt. To complete, also the Cl-/HCO3- anion exchanger AE2 and cystic fibrosis transmembrane regulator CFTR/MRP7/ABCC7 both most important for ductal bile secretion are expressed only in large bile ducts.51
Intestinal bile acid transportTo complete enterohepatic circulation an efficient reabsorption of bile acids in the intestine with subsequent delivery to portal circulation is required. Again, while parts of unconjugated bile acids may be absorbed by passive diffussion, conjugated bile acids are taken up into enterocytes by Na+-dependent and Na+-independent mechanisms.4 The first one is mediated by the apical sodium-dependent bile salt transporter Asbt, which is similar to the one also found in cholangiocytes and renal tubular cells.59 Asbt is strictly restricted to the terminal ilieum, where it solely mediates the transport of bile acids, conjugated and unconjugated primary and secondary bile acids.59 Na+-independent bile acid transport for conjugated and unconjugated bile acids is mediated by Oatp3, which in contrast to Asbt is expressed down the length of the small intestine.60 If this holds true for human OATP expression (i.e. OATP-A) in intestinal brush border membranes remains to be further evaluated.4,61 Intracellular trafficking of bile acids is mediated via the intestinal bile acid-binding protein, I-Bapb, which is cytoplasmatically attached to Asbt.62 For long it was speculated that either tAsbt58 or Mrp3 63,64 may represent the pendant to Asbt for basolateral bile acid export. However, recent findings suggest that heteromeric Osta/Ost/3, which catalyzes Na+- independent bile acid export only in its heteromeric form40,41 is the more likely candidate since it exactly mirrors Asbt distribution in intestine and kidney.41 One has to mention that also export pumps are expressed in apical brush border membranes (e.g. Mdr1, Mrp2).2 Concerning bile acid transport, Mrp2, which is highest expressed in duodenum and continuosly decreases towards colon,65,66 might play a role as alternative defense mechanism.67
Renal bile acid transportUnder normal conditions, bile acids which escape first pass effect of the liver are glomerularely filtrated and effectively reabsorbed in the proximale nephron convolute.68 In cholestasis however, this pathway can gain more importance by reduced re-uptake and enhanced active secretion.69 Similar to the terminal ilieum re-uptake is mediated by the apical located Asbt. While unconjugated and conjugated di-and trihydroxy bile acids can be transported via this salvage mechanism, sulphated bile acids are not transported, thus promoting the excretion of these compounds which are highly formed in cholestatic conditions.35 Also Oatp1 has been located to the apical membrane of the proximale tubules,70 thus potentially contributing to bile acid re-absorption. The role of Oatp3 in the kidney is still controversial.35 The contribution of active urinary bile acid secretion via apical Mrp2 and Mrp4, which are upregulated in cholestatic conditions67,71,72 has been postulated, but so far not been proven in detail. In analogy to rodents also the human counterparts have been localized in the kideny, i.e. ASBT, OATP-A, MRP2 and MRP4.59,73-75 Little is known about basolateral efflux of bile acids in the kidney. Potential candidates are Mrp3,72,76 organic ion transporters (Oat),35 or again the counterpart of Asbt on the basolateral membrane Ostα/ Ostβ, which is highly expressed in the kidney.40,41
Transcriptional regulation under physiologic conditionsThe regulation of bile acid transport systems is subject to intensive control mechanisms on transcriptional and post-transcriptional levels. On transcriptional level a complex interacting network of ligand activated nuclear (orphan) receptors (NR) and hepatic transcription factors catalyzes the physiological and pathophysiological demands. NR involved in the regulation of hepatobiliary transport systems are either directly ligand activated by distinct bile acids, steroid hormones, retinoids and a broad spectrum of diverse drugs or their nuclear availibilty and DNA binding can be modified (mainly by cytokines).8,9,11,77 Another level in mediating nuclear receptor function comprises recruitment of distinct co-activators and release of co-repressors.78 So far, three nuclear receptors, the farnesoid X receptor FXR (NR1H4), the pregnane X receptor PXR (NR1I2) and the vitamin D receptor VDR (NR1I1), have been shown to bind a distinct spectrum of hydrophobic bile acids as ligands.79-84 In addition, the liver X receptor alpha LXRa (NR1H3) was demonstrated to be activated by 6a-hydroxy bile acids and anologs.85 The constitutive androstane receptor CAR (NR1I3) is enogenously activated by bilirubin86 but a role for bile acid sensing has been postulated.87 All of these NR belong to class II nuclear receptors, that in contrast to class I steroid hormone receptors typically form heterodimers with the retinoid X receptor alpha RXRa (NR2B1) before binding to distinct DNA response elements in respective promoter regions. It is noteworthy that RXR can function as a silent heterodimer partner (e.g. for VDR, RAR) - RXR ligand binding is prevented - or as an active heterodimer partner (e.g. for FXR, LXR, PPAR)- both receptors can be synergistically activated/ modulated in the presence of both ligands.88,89 Moreover, bile acids and cytokines are able to directly affect expression levels and activity of critical hepatic transcription factors, which are essentially for hepatobiliary transporter expression.2,90-92 Rapid changes of transporter expression are mainly mediated by postranscriptional actions. An intensive regulation by second messengers and protein kinases modulates recruitment of transporters from intracellular pools to the plasma membrane and alters carrier activity by (de)/phosphorylation and protein-protein interaction processes.2,3,7 However, this review will keep its focus on transcriptional regulation and for a more detailed understanding the reader is kindly referred to recent more detailed reviews.2,6,7
FXR: The farnesoid X receptor (FXR/NR1H4) was the first and most important nuclear orphan receptor shown to be activated by bile acids.79-81 FXR is abundantly expressed in tissues belonging to the enterohepatic circulation (i.e. liver, gut) and in the kidney.93 More recent investigations found FXR also expressed in several tissues not belonging to typical bile acid target tissues (e.g. heart, thymus, ovary, eye, spleen, testis, vasculature and vasuclare smooth muscle cells).94-96 Physiological bile acids were reported to activate FXR in the rank order of potency: CDCA (chenodeoxycholic acid) > DCA (deoxycholic acid) = LCA (lithocholic acid) > CA (cholic acid).79 However, recent data suggest that LCA is an inverse agonist of FXR97 and DCA, CA and UDCA are partial FXR agonists which regulate FXR target genes in a gene-selective manner.98 Smiliar or even higher FXR binding and activity was shown for the conjugated bile acids.79,99 FXR typically forms a heterodimer with RXR and binds with high affinity to inverted repeat (IR) 1 response elements, although binding to IR0 (for SULT2A1) and everted repeats (ER) 8 (for MRP2) where also shown.100-103 One prominent exception from this rule is human UGT2B4 - glucuronidating hydrophobic bile acids-which promotor binds FXR as a monomer.104 Since bile acids represent the natural ligands for FXR activation it is not surprising that this receptor activates and represses key transporters of bile acid uptake and export as well as key enzymes of bile acid synthesis and metabolism via direct or indirect mechanisms. Both canalicular bile acid export transporter genes BSEP/ Bsep and MRP2/Mrp2 are directly transactivated via FXR:RXR binding to IR-1 or ER-8 response elements in their promotor regions, respectively.102,105-107 It is noteworthy that LCA may act as an FXR antagonist thus downregulating (stimulated) Bsep expression.97,108 While none of rodent Oatps is reported to be regulated via FXR, human OATP1B3/SLC21A8 (formerly OATP8) is directly transactivated via the IR-1 response element109 and human OATP-C is repressed (see below). Also MDR3 belong to the genes which are directly positively regulated by FXR.110 Apart from bile acid and organic anion transporters also human and murine ilieal bile acid binding protein (I-BABP/I-babp) were shown to be directly transactivated,80,111,112 further the rat bile acid bile acid-CoA: amino acid N-acetyltransferase (BAT), which taurine and/or glycine conjugates bile acids113 and bile acid sulfating sulfotransferase (STD or Sult2a1).103,114 The regulation of FXR itself in cholestasis is controversial. While there are reports that FXR is downregulated after common bile duct ligation in rats115 and CA feeding in mice,116 (Trauner & Wagner, unpublished observations), recent reports rather suggest a positive auto-regulatory loop for FXR.98,117 Repressive effects of FXR on hepatobiliary transporters are mediated indirectly via the short heterodimer partner (SHP) and/or hepatocyte nuclear factors (HNF) 4 and 1 involving bile acids as well as cytokines. The promotor of human and rodent SHP, contains FXR response elements and is positively transactivated by FXR.118 However, a negative feedback loop for this transactivation via SHP inhibition of the FXR:RXR coactivator PGC-1 seems to fine tune this mechanism.119 SHP itself, highly expressed in liver, is an unusual NR since it lacks the DNA binding domain but still contains its dimerization domain allowing to interact and negatively affect several other NR (among them HNF4, liver receptor homologue-1 (LRH-1), CAR:RXR, RAR:RXR, PXR:RXR, LXR:RXR) but positevly affects PPARa :RXR and PPARy:RXR.8 SHP mediated repression is suggested to involve competition with coactivators and its strong transcriptional repressor domain.8,120,121 So far no ligand has been reported for SHP. FXR-SHP dependent repression has been reported for rat Ntcp via repression of HNF4 and RAR:RXR binding120,122 and suggested for murine Ntcp.123 However, Ntcp downregulation upon bile acid feeding in SHP knock-out mice also seem to involve bile acid dependent, but SHP independent pathways.124 Such pathways might involve bile acid (± cytokine induction) triggered activation of the JNK pathway and reduced binding of HNF4 (proposed for rat Cyp7a1),125,126 JNK dependent phosphorylation of RXR with reduced binding of RAR:RXR to the rat Ntcp promotor,121 cytokine induction with supression of RAR:RXR, HNF1 and HNF4 (shown for rat Ntcp promoter),90,127,128 bile acid ± cytokine mediated reduced HNF4 binding (proposed for rat Ntcp and human OATP1B1 (formerly OATP-C).91,129,130 However, concerning transcriptional Ntcp regulation there might exist great species specific differences.91 It should be mentioned at this point that HNF1 is negatively regulated by HNF4 and HNF1 in turn negatively regulates its own expression and that of other HNF4 dependent genes.131 The central role of HNF1 and HNF4 as central regulators of basolateral bile acid uptake transporters is underlined by reduced Ntcp, Oatp1 and Oatp2 expression in HNF1-/- and conditional HNF4-/- mice.132,133 HNF1a was also reported to be required for expression of Oatp4.134,135 Thus, HNF1a appears to be the master regulator of basolateral Ntcp and Oatp expression. Among hepatobiliary transporters human OATP1B1 is also negatively regulated by FXR involving HNF1 and HNF4.8,91 Mouse Asbt is downregulated by bile acids involving FXR-SHP dependent reduced LRH-1 binding to the Asbt promotor,136 while human ASBT promotor is suggested to be negatively regulated via SHP dependent reduced RAR:RXR transactivation.137 The HNF1 knockout mouse also represents reduced Asbt.132 The transcriptional effects of FXR-SHP dependent and independent pathways on the regulation of CYP7A1, CYP8B1 and CYP27, involving LRH-1 and again as central regulator HNF4 should only be mentioned and the reader is referred to detailed recent reviews.2,8,18
PXR: The pregnane X receptor (PXR/NR1I2 or SXR in human) was the second NR reported to be activated by bile acids in the order of potency: 3-keto LCA, LCA, and to lesser extents by DCA and CA.82,83 One report also sug-gested UDCA as a potential ligand of PXR.138 Because of a variable ligand binding domains (LBD) there exist great differences in distinct PXR ligands between species.139,140 As such, rifampicin, which is a known human PXR activator do not activate rodent PXR while pregnenolone-16a carbonitrile (PCN) acts vice versae.141,142 Also PXR ligands itself comprise a heterogenous group including bile acids, vitamin E,143 a spectrum of several xenobiotics, including rifampicin, natural steroids or hyperforin, the active compound of St. John’s wort.141,142,144 PXR binds as PXR:RXR heterodimer to direct, indirect and everted repeats spaced by a various number of nucleotides9 and it is highley expressed in liver and intestine and to lower degrees in kidney, uterus, ovary and placenta.9,141,145 Although its primary role is determined to be xenobiotic detoxification through transactivation of human and rodent cytochrome p450 CYP3A and CYP2B isoforms77 as well as activation of the human MDR1,146,147 several recent reports recognize its capibility in modulating hepatobiliary transport systems and bile acid metabolizing enzymes (for an profiling of PXR target genes see148). So far rodent Oatp2 has been shown to be PXR dependent83 and there are also data for a positive regulating role in the transcription of MRP3/Mrp3.149,150 Together with FXR and CAR, PXR also regulates MRP2/Mrp2 expression via an common unusual ER8 element in the promotor.102 Similar to FXR, PXR represses CYP7A1 and potentially CYP8B1 via a mechanism again involving HNF4.151,152 In addition, Sult2a1114,153 and bilirubin glucuronidating Ugtiai154,155 are PXR dependent. A recent report also found a PXR binding site in the human SHP promotor156 and in turn human and murine PXR is negatively regulated by SHP.157
CAR: The constitutive androstane receptor CAR (NR1I3) has so far not been shown to bind bile acids, but to be activated by bilirubin, which accumulates in cholestatic disorders.86 Similar to PXR there is also a significant ligand dependent species variability.142 Androstane metabolites are reported to be inverse agonists, while phenobarbital is a well known CAR agonist.158 CAR again binds as CAR:RXR heterodimer to several DR and ER motifs.9 but there has also been described a binding of CAR as monomer.159 Unlike other NRs CAR is constitutively active meaning that it do not require an agonist for full activation and it is sequestered in the cytoplasm from where it translocates to the nucleus similar to classic steroid hormone receptors.8,9,58 CAR is highly expressed in the liver and intestine.160,161 CAR and PXR regulate distinct but overlapping genes invoved in bilirubin, bile acid and xenobiotic metabolism,155 part of this action may be mediated by binding of CAR and PXR to common response elements in promotor regions of CYP2B and CYP3A genes.142,162 So far CAR is shown to participate in the regulation of several Mrps, including Mrp2,102 Mrp3,65;163 Mrp4,87,150 Oatp2116,150 and bile acid and biliru-bin detoxification enzymes such as Sult2a87,114,150,164 and Ugt1a1.86,165 Again, SHP may interact indirectly with CAR via recruitment of co-repressors or inhibition of coactivators and is thus able to inhibit the transactivation of the classical CAR target gene CYP2B.166
Other nuclear receptors involved in hepatobiliary transporter regulation VDR, RAR, LRH-1, PPARa, GR): Recently also the vitamin D receptor VDR (NR1I1) was shown to be activated by LCA and its metabolites in even lower concentrations than required for PXR activation.84 The expression of VDR in hepatocytes is minimal and significantly higher in Kupffer and stellate cells and the intestine.167 While so far only bile acid detoxifying enzymes (i.e Cyp3A4/Cyp3a11, Sult2a1) have been shown to be regulated by VDR,84,114 a most recent report identified a VDR response element in the rat Asbt gene and its induction by calcitriol,168 suggesting that VDR may even play a role in bile acid transporter regulation. Moreover, FXR induced BSEP expression was also inhibited by vitamin D.169 The retinoic acid receptor RARa (NR1B1), the most prevalent isoform in hepatocytes is activated by retinoids.9 The obligate RAR:RXR heterodimers is required for constitutive expression of rat Ntcp.128 Morover, RAR:RXR binding to retinoid response elements in the promotor regions of Ntcp and Mrp2 positively transactivates both genes,128 a similar mechnism is suggested for Oatp4.135 In contrast, RARa: RXRa is suggested to negatively regulate Mrp3.170 Liver receptor homologue LRH1 (NR5A2) (also known as fetoprotein transcription factor, FTF) is involved in the upregulation of Mrp3, since a binding site for LRH-1 was found in the Mrp3 promotor.171 Since bile acids do not act as LRH-1 ligands172 another factor mediating bile acid-dependent induction might be required.171 TNFa may be this factor since it upregulates LRH-1 expression in vitro and in vivo.173 Involvement of LRH-1 has also been suggested for Asbt regulation2 and CYP8B1.174 Peroxisome proliferating activator receptor PPARa (NR1C1), activated by fibrates, statins, long chain unsaturated fatty acids and eicosanoids is involved among others (for review)9 in the regulation of MDR2,175 ASBT,176 CYP8B1177 and CYP7A1,178 UGT2B4179 and SULT2A1.180 The glucocorticoid receptor GR (NR3C1) is shown to directly transactivate human NTCP8,181 and human ASBT176 and a role in the regulation of AE2 was discussed.182 Recently and GR response element was found in the human CAR promotor.183 Most interesting, UDCA was reported to activate the glucocorticoid receptor.184
Changes in diseaseImpairment of normal bile flow leading to cholestasis may be the result of either functional defects in bile formation at the hepatocellular level or in secretion and flow at the bile duct level.6,185 Transporter alterations play an important role in the development and/or maintainance of these cholestatic disorders, but in turn may also provide anticholestatic defense mechanisms. In general, downregulation of basolateral Ntcp and Oatps, together with partially recovery of Bsep reduces hepatocellular accumulation of potentially toxic bile acids. In addition, induction of basolateral Mrp3 and Mrp4 may provide an alternative pathway for elimination of biliary compounds. Moreover, cholestasis leads to an impairment of the normal enterohepatic circulation with enhanced ductular re-uptake of bile acids via induction of cholangiocellular Asbt (“cholehepatic shunting”) and - reciprocally - reduced renal re-uptake of biliary compounds via downregulated renal Asbt, thus together with induction of renal Mrp 4 promoting bile acid elimination.2,186 Therefore, defects in transporters may be primary as the result of genetic defects or secondary acquired as a result of cholestatic injury (most forms of transporter alterations). Several animal models mimicking human intraheptic and extraheptic cholestasis have enabled us to understand the transporter alterations and pathophysiogical mechanisms on which these diseases are based. In the following, genetic and acquired human cholestatic liver disease with their corresponding animal model (mainly results from mice and rats) are confronted and general aspects are highlighted.
Genetic transporter alterationsProgressive familial intrahepatic cholestasis 1(PFIC1): PFIC1 (Byler’s disease) often begins first as recurrent and later permanent cholestasis leading to fibrosis, cirrhosis and liver failure in the first 2 decades of childhood. Typically, children represent with low y-glutamyltransferase levels and high primary bile acids in serum as well as extrahepatic manifestations (e.g. diarrhea, malabsorption and pancreatitis, hearing impairment). Histologically, a bland canalicular cholestasis without much bile duct proliferations impresses and electronmicroscopically the bile appears coarse granular in the canaliculi (Byler bile).5,187,188 PFIC1 is caused by muations in FIC1 (ATP8B1), an ami-nophospholipid flippase which is expressed in the canalicular membrane of hepatocytes and cholangiocytes.189,190 In contrast to previous suggestions191,192 FIC1 appears not to be a bile acid transporter. Thus the mechanisms, by which mutations in this gene cause a profound decrease in biliary (hydrophobic) bile acids, remains unresolved.193 Of interest, symptoms of diarrhea persist after liver transplantation, suggesting a broader role of FIC1 in disease development.2 FIC1 knock-out mice show high intestinal bile acid uptake upon bile acid feeding suggesting a defect in the regulation of intestinal bile acid absorption.194 A recent study found FXR downregulated and reduced in its activity in some PFIC1 patients which was accompanied by increased expression of ilieal ASBT and reduced expression of ilieal bile acid binding protein (ILBP).117 Similar to FIC1 knockout mice, the amount of reabsorbed bile acids in FXR knockout mice was increased, despite absence of ilieal bile acid binding protein,195 thus giving a rational for the intestinal findings in patients and mice. Moreover, reduced hepatic BSEP expression secondary to FXR alterations might account for some of the hepatic changes in these patients, although this hypothesis needs to be prooven.117 Mutations in the FIC1 gene are also associated with the benign recurrent intrahepatic cholestasis (BRIC type 1, Summerskil Syndrome), which represents with recurrent episodes of cholestasis and low y-glutamyltransferase levels and can also be associated with extrahepatic symptomes, but does not progress to chronic liver disease.5,191
Progressive familial intrahepatic cholestasis 2(PFIC2): PFIC2 (Byler’s syndrome) also do not show elevated levels of y-glutamyltransferase or bile duct proliferation but histology often presents with nonspecific giant cell hepatitis and patients are frequently jaundiced and the disease rapidly progresses to progressive cholestatis requiring liver transplantation within the first decade. In contrast to PFIC1 the bile is amorphous or filamentous and extrahepatic manifestations are uncommon5 (with the exception of cholelithiasis, wich is found in PFIC2/ BRIC2, but not in PFIC1/BRIC1).196 PFIC2 is caused by mutations in BSEP,197 thus patients only have 1% of normal biliary bile acid secretion, confirming the predominant role of this transporter for human canalicular bile acid secretion.43 Surprisingly, Bsep knockout mice only display mild nonprogressive cholestasis.44 This less severe phenotype in mice can likely be attributed to another yet unidentified canalicular bile acid transport system, which in contrast to Bsep may transport mainly hydrophilic bile acids, which are more prominent in rodents than in humans.44,198 Similar to mutations in FIC1 less severe missense mutations in the BSEP gene may also cause a form of benign recurrent intrahepatic cholestasis (BRIC type 2).196
Progressive familial intrahepatic cholestasis 3(PFIC3): PFIC3 is caused by homozygous mutations in MDR3.199 In contrast to PFIC1 and PFIC2, PFIC3 is characterized by high yGT values, bile duct proliferation and inflammatory infiltrates in portal fields.200 When the phospholipid flippase is deficient, biliary phospholipid content dramatically decreases, thus bile acids do not form mixed micelles and the lack of their protective effects on the bile duct epithelium may cause bile acid-induced injury of the biliary epithelium.199 The spectrum of MDR3 deficiency is broad ranging from neonatal cholestasis and intrahepatic cholestasis of pregnancy (ICP) to cirrhosis in adults and cholesterol gallstone formation.55,201,202 Actually, in some cases it may become difficult to distinguish between fully reversible ICP with MDR3 mutations from more serious MDR3 associated liver disease.186,203 The pathophysiological consequences of this genetic defect were established in the Mdr2 knockout mouse,48 which completely lacks biliary phospholipid levels and develop a phenotype resembling primary sclerosing cholangitis204 and spontaneously evolve cholecysto-and hepatholithiasis.205 However, patients with primary biliary cirrhosis have unchanged MDR3 mRNA levels206,207 and no MDR3 polymorphisms in PSC patients were observed.208
Dubin-Johnson Syndrome: The Dubin-Johnson syndrome is caused by mutations in MRP2 and the subse-quent loss of the canalicular transporter.209-211 This defect results in congenital conjugated hyperbilirubinemia, since MRP2 transports conjugates including bilirubin, divalent bile acids and GSH.45,46 Patients with such mutations show an overexpression of basolateral MRP3, which may represent an alternative escape pathway for otherwise accumulating biliary constituents.212 Similar, groningen yellow (GY/TR-) wistar rats and Eisai hyperbilirubinemic Sprague Dawley rats (EHBR) lack Mrp2 expression and have chronic conjugated hyperbilirubinemia with reduced bile flow and adaptive overexpression of Mrp3.63,213-215
Genetic defects in basolateral bile acid transporters have so far not been reported, although OATPs appear to be highly polymorphic.3,216 Animal models with Mrp3 and Mrp4 deficiency exist.87,217 However, while first results from cholestatic Mrp4 knockout mice are yet not published, bile duct ligation in Mrp3 knockout mice confirmed the only minor role of intestinal Mrp3 for enterohepatic bile acid circulation (see Osta/3), the importance of this transport system for regurgitation of conjugated bilirubin from hepatocytes into blood, but surprisingly no protective effect of Mrp3 on liver injury or bile acid levels.217
Acquired transporter alterationsBland cholestasis: Non inflammatory, bland cholestasis can evolve as drug-induced intrahepatic cholestasis (for review218) or as intrahepatic cholestasis of pregnancy (ICP) (for review.219-221) Typical targeted transporters of drug associated cholestasis are BSEP and MRP2. Cyclosporin A induce disorganization of BSEP localization resulting in its intracellular relocation and disruption of the pericanalicular F-actin cytoskeleton222 and estradiol-17/3-D-glucuronide (as modell for oral contraceptive-induced cholestasis and ICP) induces endocytotic internalization of Bsep and Mrp2, thus impairing bile acid dependent-and independent bile flow.223,224 Also rifamycin SV, rifampicin, glibenclamide and troglitazone cis-inhibit Bsep/BSEP mediated taurocholate transport, thus providing a mechanism for intrahepatic cholestasis caused by these agents.3 In addition, the endothelin antagonist bosentan inhibits BSEP/Bsep but induce Mrp2 mediated biliary bilirubin excretion and bile acid independent bile flow.225,226 The development of ICP is multifactorial including potential hepatobiliary transporter affections, sex hormonal maladaptations, dietary and environmental factors. ICP affects up to 2% of pregnancies, has a recurrence rate of 40 to 60%, typically starts with pruritus on palms and soles, and primary bile acids, which are 10 to 100 times higher than in normal pregnancies. The symptoms usually disappears a few days after delivery.220 While genetic variations in MDR3 (see also PFIC3) may be involved in the pathogenesis of ICP, recently analyzed BSEP sequence variations indicate that this gene might be of minor importance.227 Also estrogens may contribute to intrahepatic cholestasis of pregnancy and can cause oralcontraceptive-induced cholestasis.185,228,229 In the ethinyl estradiol (EE) fed rodent model Mrp2 and Bsep protein is reduced, however, Bsep expression is restored in a longstanding model of EE treatment.230 These alterations of canalicular transporters is accompanied by a reduction of bile flow of 40%.231 In addition, Na+-dependent (Ntcp) and Na+-independent (Oatp1) bile acid uptake transporters are also downregulated in this model.232 EE induced SHP expression might be involved in this downregulation (see above and ref 233). Of note, different baseline expression levels of transporters between genders and in pregnancy may also contribute to a gender specific disease development or the appearance of a specific disease in pregnancy. As such it was shown that Ntcp was either downregulated in pregnant rats and mice involving downregulation of HNF1 and reduced RAR:RXR binding to the promotor234,235 or it was upregulated in postpartum rats involving prolactin mediated Stat5 binding to the Ntcp promotor.236 Also Oatp2 and Mrp2, Mrp3 and Mrp6 are downregulated in pregnant rats, while in contrast Mrp2 was upregulated in pregnant mice, suggesting species differences under this circumstances.235-237 Of interest, PXR and its target gene Cyp3a11 were reported to be higher in pregnant mice238 and CAR to be higher in human females compared to males.239 Differences between genders were also reported for Oatp1 and Bcrp (higher in females) and Mdr1a and Mrp4 (higher in males) so far.235,240
Inflammatory cholestasis: Clinically, this form of cholestasis is frequently associated with sepsis in the course of intra-and extrahepatic infections, drug-and alcohol induced liver injury, total parenteral nutrition, neoplastic disorders and postoperatively. The common pathophysiological mediator in this entity is endotoxin (lipopolysaccharides (LPS) of the outer bacterial membrane) and/or the release of proinflammatory cytokines (e.g. tumor necrosis factor a (TNFa), interleukin 1/3 (IL1/3)).241 In liver biopsies from patients with inflammation-induced cholestasis profound reductions of basolateral NTCP and OATP2 and canalicular BSEP and MRP2 have been observed.242 In the experimental model LPS reduces hepatobiliary transporters at the basolateral and the canalicular membrane, thereby reducing bile acid dependent and independent bile flow. Similar effects were demonstrated for TNFa and IL1/3.243-247 LPS leads to a dramatic decrease of Ntcp.127,248 The mechanisms involved in this downregulation include reduced binding of HNF1 and RXR:RAR to the rat promotor,127,128,249 decline of RXRa, -/, -Y mRNA,250 phosphorylation with reduced activity of RXR via mitogen-activated protein kinase 4 and JNK120,m and reduced HNF4 and 1 activity.127,129 SHP does not appear to be invoved in LPS mediated Ntcp downregulation.123 Of note, Kupffer cell depletion as well as anti-TNFa and IL1/ strategies prevent supression of Ntcp in endotoxin-treated rats and mice.251,252 Endotoxin mediated downregulation of the Na+-idependent bile acid uptake transporters Oatp1,2,4 involves reduced expression of HNF1,129,253,254 reduced expression of PXR and CAR, at least for Oatp283,250,255 and, at least for Oatp4 decreased binding activities of HNF1, RXR:RAR, C/EBP and HNF3.135 Bsep expression is only modestly decreased in inflammation-induced cholestasis upon LPS-treatment by 30%,230 while Mrp2 is profoundly reduced256 by mechanisms including repressed retinoid transactivation via RXR:RAR,128 retrieval from the canalicular membrane,257 phosphorylation with subsequent nuclear export of RXR121 or decreased activity of nuclear receptors FXR, PXR and CAR.258-261 Mrp3 expression was shown to increase TNFa dependent via upregulation of LRH-1173 and after LPS treatment,262,263 however, there are also reports demonstrating downregulation of Mrp3 in response to LPS and TNFa.264
Obstructive cholestasis: Obstructive cholestasis in man may appear as intraluminal obstruction (e.g. gall-stones, neoplasias), extraluminal compressions (e.g. neoplasias) biliary atresia, vanishing bile duct syndrome (e.g. primary biliary cirrhosis (PBC) with destruction of small and medium bile ducts) and primary sclerosing cholangitis (PSC) with affection of intra-and extrahepatic large ducts. In patients with extrahepatic biliary atresia NTCP mRNA levels were decreased and subsequently increased if complete biliary drainage by portoenterostomy (Kasai procedure) was achieved265 while BSEP expression was well preserved.266 In patients with biliary obstruction prior to percutaneous transhepatic biliary drainage (PTBD) MRP2, BSEP and MRP3 was reduced.267 Transporter changes in PBC are disease stage dependent: no changes of NTCP, OATP2, BSEP, MDR3 and MRP3 are observed in stage I and II,242 while in the more advanced stages III and IV NTCP and OATP2 are downregulated and MRP3 and MDR1 and 3 are induced.207 This downregulation of import and upregulation of export mechanisms may account for the shift towards renal bile acid elimination in PBC and the increase in bilirubin and serum bile acids in these stages.69 BSEP and MRP2 are transiently induced in PBC stage III, potentially aiming to overcome bile acid burden, but return to baseline levels in end stage disease IV.207 In PSC patients however, MRP2 was downregulation, while OATP-A is up-regulated in contrast to downregulation of OATP2.268 One widely used model for obstructive cholestasis is common bile duct ligation (CBDL) in rodents. In these models hepatic uptake systems (i.e. Ntcp, Oatp1) are downregulated,269,270 while canalicular export pumps, Bsep and Mrp2 in mice are maintained.271 Of note, in rats CBDL results in a marked reduction of Mrp2.256 In addition, basolateral alternative efflux pumps (i.e. Mrp3 and Mrp4) are overexpressed,56,271-273 which may shift bile acids and bilirubin from hepatocytes into the systemic circulation. Overex-pression of potential renal bile acid export transporters Mrp2 (in rats)71 and Mrp4 (in mice),72 with concomittant reduction of renal re-uptake systems (i.e Asbt),71 should enhance alternative elimination via urine. Induced canalicular Asbt may mediate reuptake of bile acids from the obstructed biliary lumen.71 Mechanism underlying these transporter changes may consist of an inflammatory component since CBDL results in induction of proinflammatory cytokines274-276 and the accumulation of endogenous bile acids and biliary compounds (e.g. bilirubin) which activate several nuclear receptors (e.g. FXR, PXR, VDR, CAR). Activation of FXR by accumulating bile acids induces SHP and may downregulate Ntcp.123,252 Alternatively, it has been shown that bile acids are also capable of inducing SHP via an FXR-independent mechanism involving JNK.126 Bile acids have also been shown to suppress HNF4 transcription,90,91 which might reduce HNF1 and subsequently Ntcp. Systemic proinflammatory cytokines have no effect on Ntcp expression during obstructive cholestasis since TNFa and IL-1/3 antagnosits have no effects on Ntcp expression.252 However, Mrp2 downregulation in rats was attributed to cytokine dependent inhibition of RAR:RXR binding to the Mrp2 promotor.277 TNFa induced LRH-1 as well as reduced RAR:RXR might be involved in the upregulation of Mrp3.170,173 In addition activation of PXR and CAR by bile acids and/or bilirubin might contribute to the observed changes.149,163 Similar mechanisms may play a role in the induction of Mrp4 via CAR.86,87 The Mdr2 knockout mouse represents an excellent model for studing sclersoing cholangitis204,278 and long term effects of cholestasis on transporter expression. Mdr2 knockout mice show persistent downregulation of Ntcp, but this is obviously not sufficient to prevent intrahepatic bile acid accumulation. Moreover, Mdr2 knockout mice do not show adaptive upregulation of alternative basolateral Mrp3 and Mrp4 expression when the cholestatic phenotype is fully developed, although intrahepatic bile acid levels remain increased.279 A model for non surgical intrahepatic reversible cholestasis is administration of a-naphtylisothiocyanate (ANIT).280,281 A recent study using rats found a common cholestatic transporter pattern with downregulation of Ntcp and Oatp1, while Bsep remained unchanged and Mdr2 as well as Mrp3 were upregulated.282
Therapeutic options 2005 and beyondCounteracting cholestatic disorders should be a multilevel approach to target impaired physiological functions and pathophysiological mechanisms of bile acid/bilirubin transport and metabolism and to create/support adaptive rescue pathways for potential toxic accumulating biliary compounds. Unraveling the mechanism and “players” lying behind physiological and pathophysiological cholestatic conditions allows a better understanding of empiric treatment strategies (i.e. UDCA, rifampicin (Rifa), Phenobarbital (PB)) and the development of novel, more effective treatment options customized to the special requirements of the proper cholestatic disorder (Table II).
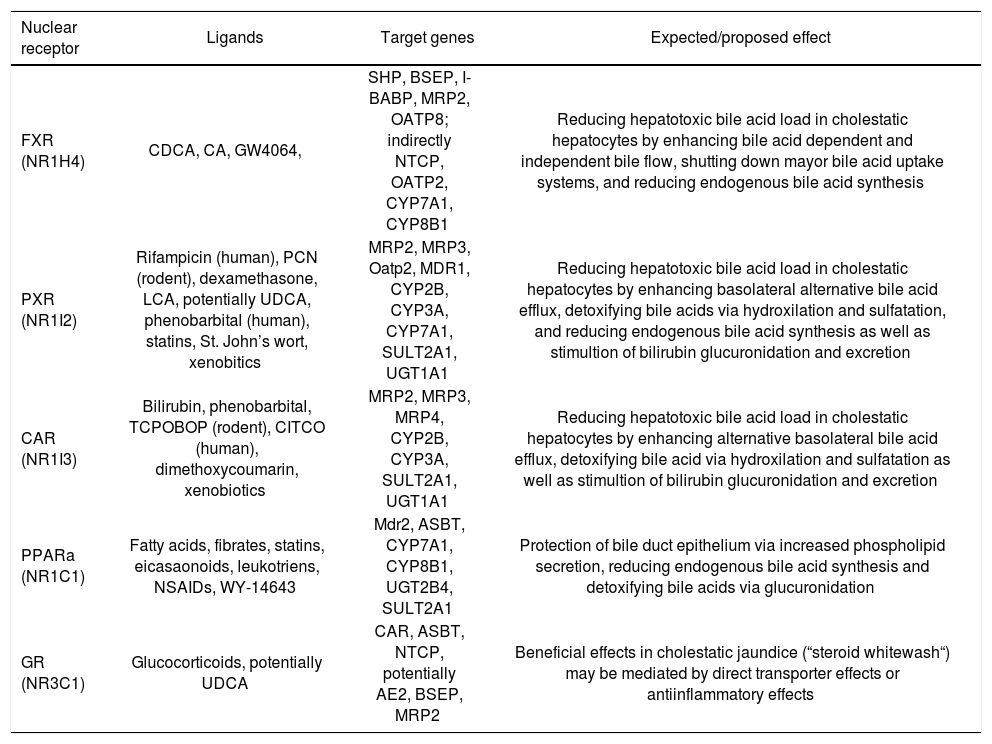
Nuclear receptors agonists and their expected effects in cholestatic liver disease.
| Nuclear receptor | Ligands | Target genes | Expected/proposed effect |
|---|---|---|---|
| FXR (NR1H4) | CDCA, CA, GW4064, | SHP, BSEP, I-BABP, MRP2, OATP8; indirectly NTCP, OATP2, CYP7A1, CYP8B1 | Reducing hepatotoxic bile acid load in cholestatic hepatocytes by enhancing bile acid dependent and independent bile flow, shutting down mayor bile acid uptake systems, and reducing endogenous bile acid synthesis |
| PXR (NR1I2) | Rifampicin (human), PCN (rodent), dexamethasone, LCA, potentially UDCA, phenobarbital (human), statins, St. John’s wort, xenobitics | MRP2, MRP3, Oatp2, MDR1, CYP2B, CYP3A, CYP7A1, SULT2A1, UGT1A1 | Reducing hepatotoxic bile acid load in cholestatic hepatocytes by enhancing basolateral alternative bile acid efflux, detoxifying bile acids via hydroxilation and sulfatation, and reducing endogenous bile acid synthesis as well as stimultion of bilirubin glucuronidation and excretion |
| CAR (NR1I3) | Bilirubin, phenobarbital, TCPOBOP (rodent), CITCO (human), dimethoxycoumarin, xenobiotics | MRP2, MRP3, MRP4, CYP2B, CYP3A, SULT2A1, UGT1A1 | Reducing hepatotoxic bile acid load in cholestatic hepatocytes by enhancing alternative basolateral bile acid efflux, detoxifying bile acid via hydroxilation and sulfatation as well as stimultion of bilirubin glucuronidation and excretion |
| PPARa (NR1C1) | Fatty acids, fibrates, statins, eicasaonoids, leukotriens, NSAIDs, WY-14643 | Mdr2, ASBT, CYP7A1, CYP8B1, UGT2B4, SULT2A1 | Protection of bile duct epithelium via increased phospholipid secretion, reducing endogenous bile acid synthesis and detoxifying bile acids via glucuronidation |
| GR (NR3C1) | Glucocorticoids, potentially UDCA | CAR, ASBT, NTCP, potentially AE2, BSEP, MRP2 | Beneficial effects in cholestatic jaundice (“steroid whitewash“) may be mediated by direct transporter effects or antiinflammatory effects |
NOTE. CA, cholic acid; CDCA, chenodeoxycholic acid; CITCO, 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-ichlorobenzyl)oxime; 6-ECDCA, 6-ethyl chenodeoxycholic acid; LCA, lithocholic acid; NSAIDs, non steroidal antiinlammatory drugs; PCN, pregnenolone 16a-carbonitrile; TCPOBOP, 1,4-bis-[2-(3,5-dichlorpyridyloxy)]benzene; UDCA, ursodeoxycholic acid.
Ursodeoxycholic acid: To date UDCA is the only approved drug for treatment of PBC and PSC, since UDCA was shown to improve biochemical and clinical parameters in a variety of cholestatic syndromes and positively effects survival free of liver transplantation in PBC.283,284 The mechanisms of action of UDCA are multi-level comprising transcriptional transporter induction and targeting of bile acid metabolizing enzymes (see below), posttranscriptional transporter trafficking followed by increased insertion of canalicular transport systems (Bsep) into the canalicular membrane of rats and stimulating of transporter function by inducing phosphorylation of ABC 285 while additionally converting a hydrophobic bile acid pool into a more hydrophilic one (for review).283,284,286 Most of the knowledge however, is obtained from experiments in animals. Albeit UDCA changes transcriptionally gene expression,67,287 no definite nuclear receptor has been elucidated. Of note, FXR and PXR may mediate some of the UDCA actions,98,138 but most of the transcriptional transporter effects are FXR independent.67,271 UDCA stimulates bile acid dependent and independent bile flow in naïve mice via induction of canalicular Bsep and Mrp2.67,287 This “orthograde” biliary excretion is extended by induction of additional adaptive basolateral efflux pumps Mrp3 and Mrp4, which are suggested to efflux bile acids “retrogradely” back into the systemic circulation followed by their renal elimination.67,288 Stimulation of renal (Mrp2, Mrp4) and intestinal (Mrp2, Mrp3) bile acid efflux pumps might support the elimination of potentially toxic biliary constituents in other bile acid targeted tissues, thus contributing to an overall enhanced bile acid elimination.67 While parts of these adaptive transporter alterations are already intrinsically induced in cholestatic animal models72,271 (Tauro)UDCA failed to additionally stimulate these transporters to sig-nificant extents in cholestatic mice282 (and Wagner and Trauner, unpublished observations) demanding the use of more potent therapeutical agents. In healthy humans UDCA induces BSEP and MRP4 expression on posttranscriptional levles.289 In PBC UDCA enrichment correlated positively with OATP1B1 and MRP2,207 thus potentially contributing to reduced bilirubin and bile acid levels observed in these patients.283,286 On metabolic levels UDCA has been shown to stimulate bile acid hydroxylating CYP3 A4/Cyp3 all in human and rodents138,290 and to downregulate human CYP7A1 the key enzyme of bile acid synthesis in vitro.98,291 Thus, UDCA appears to act on transporter level by restoring defective transporters (e.g. Bsep, Mrp2) and generating alternative overflow-systems (Mrp3, Mrp4) for accumulating biliary compounds and on metabolic level by induction of detoxification pathways (Cyp3a11).
FXR agonists: Pharmaceutical compounds targeting FXR are promising therapeutical approaches since studies in humans and rodents revealed a predominant role of disrupted FXR in diverse aspects of cholestasis related disorders. As such FXR was reported to be downregulated upon CA challenge,116 endogenous bile acid load in CBDL115 and in response to proinflammatory cytokines (TNFa and IL-1) and endotoxin.258 PFIC1 is associated with markedly reduced FXR levels117 and PFIC2 patients lack the typical primary FXR target BSEP.43 Moreover, FXR-- mice exhibit severe liver injury and mortality after CA feeding,67,112 which could be explained by the lack of downregulation of the bile acid uptake systems Ntcp and export pump Bsep as well as the lack in shutting down endogenous bile acid formation via Cyp7a1 and Cyp8b1.112 However, CBDL in FXR-- did not result in aggravation of liver injury.271 This effect may be attributed in part to the induction of FXR independent alternative hepatic (Mrp3, Mrp4) and renal (Mrp2, Mrp4) export pumps and induction of bile acid detoxification which are higher in FXR-- (Cyp3a11 and Sult2ai).72,108,138,271,292 FXR agonists, thus should overcome cholestatic disorders with reduced bile flow via stimulation of Bsep (increasing bile acid-dependent bile flow) and Mrp2 (increasing bile acid-independent bile flow), reducing bile acid uptake by downregulating Ntcp and Oatp1 and bile acid formation via reduction of Cyp7a1 and Cyp8b1.67,97,102,112,270,291,293 Induction of MDR3 via FXR might protect the bile duct by increasing the biliary phospholipid content.110 However, in the presence of biliary obstruction a choleretic dose of UDCA (a partial FXR agonist, which induces Bsep)98 in CBDL mice results in increased biliary pressure with rupture of cholangioles resulting in aggravation of bile infarcts and an increased mortality.204 Unfortunately, many clinically relevant chronic cholestatic liver diseases such PSC have a significant obstructive component, thus lowering the therapeutical use of FXR agonists in advanced disease stages. First experiments with synthetic FXR agonists in cholestatic rodent models have been promising and resulted in reduced biochemical and histomorphological markers of liver injury in some models.282,294,295 Noteworthy are also the antifibrotic effects of the strong halfsynthetic FXR agonist 6-ECDCA in vitro and in vivo, showing that FXR may be a negative regulator of hepatic stellate cells via SHP and the AP-1 transcription factor complex.296 However, the therapeutical use of FXR agonists could be limited because of interfering regulatory functions of FXR in lipid homeostasis, particularly in HDL and triglyceride metabolism.293
PXR and CAR agonists: In contrast to FXR which is downregulated under cholestatic conditions115,116 (and Wagner, Trauner unpublished observations) PXR and CAR remain almost unchanged after bile acid treatment116 thus providing ideal therapeutical targets in cholestsis. Long before knowing their exact mode of action PXR (rifampicin) and CAR (phenobarbital) ligands have been used for treatment of cholestatic disorders and symptoms (jaundice, pruritus). However, in most cases effects on cholestatic parameters in PBC were only minor.297-304 The potent rodent PXR ligand PCN was reported to protect against LCA induced liver injury on various levels, effects, which may be mediated by induction of Cyp3a11,82 induction of Oatp2 and downregulation of Cyp7a1,83 moreover by induction of bile acid sulfating sulfotransferase (Sult2a1) together with its sulfonyl donor 3'-phosphoadenosine 5'-phosphosufate synthetase 2 (PAPSS2).153 In addition, PXR activation might contribute to reduced injury by its effects on bilirubin glucuronidating Ugt1a1 and exporting Mrp2.102,154 Similar to FXR stimulation it was suggested that PCN also has antifibrotic effects by reducing a-smooth muscle antigen and collagen as fibrotic markers.305
In addition to phenobarbital as prototypic CAR activator certain compounds of traditional Chinese herbs, which have long been used for treatment of neonatal jaundice in humans were found to be potent CAR activators.165 Stim-ulation of CAR in mice drastically reduced serum levels of infused bilirubin via induction of bilirubin uptake (Slc21a6), glucuronidation of bilirubin (Ugt1a1) and canalicular excretion via Mrp2.86 In mouse models of cholestasis CAR agonists were shown to defend LCA induced toxicity primarily by induction of Sult2a and PAPSS2 gene expression with subsequent LCA sulfatation and of minor importance again via induction of Cyp3a11 with subsequent bile acid hydroxylation.164 Also the stimulatory effects of CAR on Mrp3 and sulfated bile acid transporting Mrp4 together with coordinated induction of Sult2a1 might contribute to the beneficial effects of CAR ligands.87,163 This fundamental role of CAR in bile acid detoxification is further strengthened by a study exploring CA toxicity in FXR- - and PXR--, which revealed that the use of CAR agonists can mitigate CA induced toxicity even when both classical bile acid receptors are knocked-out.116 In summary, the use of specific agonists for PXR or CAR should counteract hepatotoxicity of accumulated bile acids and bilirubin by a orchestra of hydroxilation (via Cyp3a11 and potentially Cyp2b10), sulfatation (via Sult2a1and PAPSS2) and glucuronidation (via Ugt1a1) reactions as well as coordinated induction of transporters (Mrp3, Mrp4) for these substrates. However these drugs may also toxify certain previously untoxic compounds to their toxic forms,163,306 enhance metabolism of other adminstered drugs or may interact endogenous metabolic pathways.307 Moreover, it should be noted that the liver tumor promoting potential of PB is directly linked to the presence of CAR308 and that PXR might play a role in the biology of endometrial cancer.309
PPARa agonists: The rational for the therapeutic use of PPARa agonists in cholestasis is given by the observations that fibrates or statins, both being ligands for PPARa showed beneficial effects on biochemical parameters of cholestasis and/or transaminases.310-313 The beneficial effects could at least in part be explained by the potential to stimulate canalicular phospholipid flipping via Mdr2 and thus protecting the bile duct epithelium from detergent effects of bile acids by enhanced biliary phospholipids secretion and formation of mixed micelles.175,314-317 In addition, repression of CYP7A1 (so far only shown for fibrates),318 or pleiotropic anti-inflammatory effects may contribute.319Corticosteroids: The beneficial effects of corticosteroids (e.g. prednisolone or budesonide), which have been used in acute cholestatis (e.g., “steroid whitewash” in drug-and inflammation-induced cholestasis) as well as chronic choestatic disorders such as PBC320-322 could be attributed to their anti-inflammatory and immune-modulatory effects and, at least in part, to effects mediated via transporter and enzyme alterations. As such, corticosteroids were reported to induce Mrp2 and Bsep and counteract its downregulation after endotoxin treatment in vitro.257,323 These effects could possibly mediated by targeting the glucocorticoid receptor, although this has so far only been shown for intestinal ASBT324 and NTCP.181
In summary, targeting nuclear receptors with specific potent agonists is a promising new therapeutic option for modulating distinct bile acids and/or bilirubin pathways. The development of gene-selective agonists will potentially allow the modulation of single transporter and detoxification systems depending on the given requirements in future.
Summary and outlookHereditary and acquired alterations in hepatobiliary transporter systems may result in the development or maintainance of cholestatic disorders. On the other hand adaptive induction of overflow and detoxification systems by the affected hepatocyte results in the primary intrinsic attempt to counteract liver injury. However, in chronic cholestasis these adaptions are not sufficient. The increasing information on the molecular mechanisms of bile acid transport and metabolism under physiological and pathophysiological conditions may guide the development of novel, more effective drugs. Nuclear receptors and their target genes are promising treatment options for cholestatic liver diseases.



