



Hepatic fibrosis is characterized by the accumulation of extracellular matrix which includes the accumulation of α-smooth muscle actin (α-SMA), collagen type I (COL1α1), as well as remodeling induced by metalloproteinases and tissue inhibitor of metalloproteinase (TIMPs), where hepatic stellate cells (HSCs) play a central role. In addition, the transcription factor SNAI1 (which participates in epithelial-mesenchymal transition, EMT) and mitofusin 2 (MFN2, a mitochondrial marker) plays an important role in chronic liver disease. Turnera diffusa (TD), a Mexican endemic plant, has been shown to possess antioxidant and hepatoprotective activity in vitro. We treated human HSC (LX2 cells) with a methanolic extract of Turnera diffusa (METD) to evaluate the mechanism involved in its hepatoprotective effect measured as fibrosis modulation, EMT, and mitochondrial markers.
Materials and methodsHSC LX-2 cells were treated with METD (100 and 200ng/mL) alone or combined with TGF-β (10ng/mL) at different time points (24, 48, and 72h). α-SMA, COL1α1, MMP2, TIMP1, SNAI1, and MFN2 mRNAs and protein levels were determined by real-time quantitative PCR and Western Blot analysis.
ResultsWe found that METD decreases COL1α1-mRNA, α-SMA, and TIMP1 protein expression in LX2 cells treated with and TGF-β. This treatment also decreases MFN2 and TIMP1 protein expression and induces overexpression of MMP2-mRNA.
ConclusionsOur results suggest that a methanolic extract of Turnera diffusa is associated with an antifibrotic effect by decreasing profibrotic and mitochondrial markers together with the possible induction of apoptosis through SNAI1 expression in activated HSC cells.
transforming growth factor-beta
hepatic stellate cells
extra cellular matrix
reverse transcriptase – quantitative polymerase chain reaction
methanolic extract of turnera diffusa
The liver can be damaged by a variety of acute, chronic, or intermittent acute insults that in turn induce progressive fibrosis, demonstrated as inflammatory damage, extracellular matrix (ECM) deposition, death of parenchymal cells, and angiogenesis [1]. During liver damage, the liver parenchyma undergoes necrosis and/or apoptosis; the release of cellular contents and reactive oxygen species (ROS) activate hepatic stellate cells (HSCs) and macrophages. HSCs secrete several soluble factors, including cytokines, chemokines, and growth factors, including transforming growth factor-β (TGF-β), which is the main profibrogenic cytokine and stimulates transdifferentiation of HSC to myofibroblasts. Activated macrophages release platelet-derived growth factor (PDGF) that stimulates the proliferation of myofibroblasts, and these continue to produce TGF-β, perpetuating transdifferentiation, the production of α-smooth muscle actin (α-SMA), collagen type I (COL1α1) matrix metalloproteinases (MMPs), and tissue inhibitor of metalloproteinase (TIMPs), resulting in progressive deposition of ECM and accumulation of tissue damage [2].
In a fibrotic process, changes in the ECM are driven by modulation of MMPs and TIMPs expression. The three most important MMPs expressed in liver injury and fibrosis are MMP2, MMP9, and MMP3 [3]. TIMPs control several functions, which overlap but have different inhibitory profiles for MMPs. TIMP-1 inhibits the active forms of MMP2, MMP7, and MMP9, and its overexpression has been associated with progression to cancer [4].
TGF-β has other pleiotropic functions. It induces the activation of snail family transcriptional repressor 1 (SNAI1), a transcription factor that regulates subsequent processes such as apoptosis and epithelial-mesenchymal transition (EMT) [5].
Also, several chronic liver diseases are associated with early initiating events of mitochondrial damage and dysfunction [6]. Mitochondria are a highly dynamic organelle in constant fission and fusion, and the balance of processes regulates its morphology and normal function [7]. In mammals, the fusion of mitochondria is regulated by two mitofusins, MFN1 and MFN2. In addition to the fusion role, MFN2 participates in mitochondria-mitochondria interaction and the juxtaposition of mitochondria with other organelles, particularly with the endoplasmic reticulum (ER), and its expression has been related to chronic liver disease [8,9].
Currently, some agents stimulate and favor different processes involved in liver regeneration with the consequential reversal of fibrosis. Antifibrotic agents that stimulate liver regeneration more efficiently are still being sought. Since ancient times, plants have been an important source of bioactive compounds against various diseases [10]. Traditional herbal medicine and the use of natural compounds to treat diseases are widely used in society. One of our approaches is the systematic search for natural compounds that show scientific evidence of activity or hepatoprotective properties in vitro. We have evaluated several plants from northeast Mexico to determine their usefulness for liver disease treatment [11]. One of these endemic plants, Turnera diffusa (TD) has been shown to possess antioxidant and hepatoprotective activity in vitro. We demonstrated that HepG2 cells treated with CCl4 and further exposed to TD extracts showed decreased levels of AST [11]. Furthermore, we isolated a compound, named as hepatodamianol (flavonoid C-glycosylated derived from luteolin) being the main compound responsible for this activity, that showed a maximum enzyme decrease effect in exposed cells [12,13].
Based on this, we aimed to study the potential mechanism(s) by which treatment with a methanolic extract of Turnera diffusa (METD) decreases liver injury enzymatic markers in HEPG2 cells, by using an in vitro model of a human hepatic stellate (HSC) cell line, LX-2, exposed to METD and evaluating modulation of fibrosis, EMT, and mitochondrial markers.
2Material and methods2.1Preparation of a methanolic extract of Turnera diffusa (METD)All solvents used in this study were analytical grade, except HPLC-grade methanol (Fisher Scientific, Fair Lawn, NJ). To obtain a flavonoid-rich subfraction we worked with an extract of the aerial part of TD (stems and leaves); subsequently, chlorophylls were removed by solid-phase extraction (phase C-18 cartridge, 1000mg/8mL; Alltech). The extracted product was eluted subsequently with aqueous methanol 50%, 70%, and finally, 100% methanol. The fraction obtained with 50% aqueous methanol was fractionated using silica vacuum column chromatography (Silica 60G, Merk Millipore); it was then eluted with a mix of methylene chloride, ethyl acetate, ethyl acetate:methanol 1:1 and methanol. The subfraction obtained with ethyl acetate:methanol 1:1 was the one tested in this work. It was characterized by quantification of hepatodamianol (flavonoid C-glycoside) in a Waters Alliance 1525 liquid chromatography system (Waters, USA) equipped with an online degasser, a binary pump, autosampler, and a 2996 diode array detector (HPLC-DAD) (Waters, USA). Separation was carried out in an inverse phase HypersilGold column (150mm×4.6mm, 5μm, Thermo Fisher) under the conditions previously established [12].
2.2Cell cultureWe used an in vitro model of human hepatic stellate cells (LX-2) kindly donated by Dr. María Luz Martínez Chantar (Metabolomics laboratory, CIC bioGUNE, Spain). Cells were cultured in Dulbecco's Modified Eagle medium (DMEM; Gibco, Grand Island, NY, USA), supplemented with 2% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA), 2mM L-glutamine, 100U/mL penicillin, and 100ug/mL streptomycin, and incubated at 37°C with a humid atmosphere containing 5% CO2.
2.3Cell viability assayLX-2 cells (4×103cells/well) were seeded in 96-well plates in growth medium containing serum, and after 24h, cells were treated with 0.1% DMSO (Sigma–Aldrich, Saint Louis, MO, USA) and/or METD (10 to 0.05mg/mL) for 24, 48, and 72h. Following incubation, cell viability was evaluated using an MTT [3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (Roche Diagnostics GmbH Mannheim, Germany), according to the standard experimental protocol. Cell viability was calculated comparing results from METD-treated cells with untreated cells (100% viability).
2.4HSC exposed to METD treatmentSince METD was dissolved with 0.1% DMSO, we used the same amount of DMSO in all treatments as an untreated control. Human HSC (LX2) were seeded in complete DMEM for 24h, then cells were treated with a different amount of METD (100 or 200ng/mL) alone or combined with TGF-β1 (10ng/mL) (Peprotech, Rocky Hill, NJ) at different times (24, 48, and 72h) in FBS-free DMEM.
2.5RNA extraction and Retrotranscription assayLX-2 cells were seeded at 1×105cells/well in a 24-well plate with complete DMEM for 24h. The different treatments were added in FBS-free conditions. Cells were harvested at each time point (24, 48, and 72h) and total RNA was extracted using TRIzol reagent (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer's specifications. RNA was precipitated with 100% isopropanol, washed with 75% alcohol, resuspended in 12μl RNase-free water, and then stored at −80°C. Subsequently, 300ng of RNA was used for cDNA synthesis with the MML-V RT-enzyme (Life Technology; Thermo Fisher Scientific, Carlsbad, CA, USA) as described by the manufacturer.
2.6Quantitative PCR (qPCR) for fibrosis markersWe used 200ng of cDNA to perform qPCR to quantify mRNA levels for α-SMA, COL1α1, MMP2, TIMP1, SNAI1, MFN2, and β-actin-mRNA as an endogenous control. The real-time PCR thermocycler used was a StepOne Plus (Applied Biosystems, Foster City, CA, USA). SYBR green was used for detection, and the following primers were used: α-SMA forward (+) 5’-CTACTGCTGAGCGTGAGATTG-3, α-SMA reverse (−) 5-CAGGCAACTCGTAACTCTTCTC; COL1α1 (+) 5-CGATGGATTCCAGTTCGAGTATG-3, COL1α1 (−) 5-CTTGCAGTGGTAGGTGATGTT-3; MMP2 (+) 5-GACAGGTGATCTTGACCAGAAT-3, MMP2 (−) 5-GTGTGTAGCCAATGATCCTGTA-3; TIMP1 (+) 5-CAATTCCGACCTCGTCATCAG-3 and TIMP1 (−) 5-CCTAAGGCTTGGAACCCTTTATAC-3, SNAI1 (+) 5-CCACGAGGTGTGACTAACTATG-3, SNAI1 (−) 5-ACCAAACAGGAGGCTGAAATA-3 and MFN2 (+) 5-CCTTCCTTGAAGACACGTACAG-3, MFN2 (−) 5-GATGCCTCTCACTTTGGATAGG-3. For each qPCR reaction, the following reagents were used: 10μL of SYBR-Green PCR Master Mix 2x (Applied Biosystems, Foster City, CA, USA), 200–400nm of each primer, 200ng of cDNA, completing a total volume of 20μL. Thermal cycling conditions were 95°C for 10min, followed by 40 cycles of 95°C for 15s and 60°C for 60s. The TaqMan assay was used for β-actin (Applied Biosystems, Foster City, CA, USA), according to the manufacturer's specifications. β-actin expression was used as a housekeeping gene and the fold changes of gene expression were calculated by the 2−ΔΔCt method.
2.7Protein expression levels of fibrosis markersLX-2 cells were seeded at 3×105cells/well in a 6-well plate with complete DMEM. The different treatments were added 24h later in the absence of FBS. Then, total protein extraction was performed at different time points (24, 48, and 72h), with 1X lysis buffer containing 10mM Tris-HCl (pH 7.5), 50mM KCl, 2mM MgCl2, 1% Triton X-100, 1mM dithiothreitol, 1mM phenylmethylsulfonyl fluoride, and Complete Protease Inhibitor Cocktail, according to the manufacture conditions (Roche, Mannheim, Germany), and then quantified by the Bradford method (Bio-Rad, Hercules, CA, USA). We used 50μg of protein for 12% SDS-PAGE gels and transferred them to PVDF membranes (Amersham Biosciences, Freiburg, Germany). Membranes were incubated with the following antibodies, anti-α-SMA (ab32575, Abcam, Cambridge, MA, USA) at a dilution 1:1000, anti-TIMP1 (ab109125, Abcam) at a dilution 1:1000, MFN2 (Ab56889, Abcam) at a dilution 1:1000 and anti-GAPDH (MAB5718, R&D Systems, Minneapolis, MN, USA) at a dilution of 1:2500. Membranes were washed with TBS-Tween, then incubated with horseradish-peroxidase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Promega, Madison, WI, USA) both at a dilution 1:10,000. Signal detection was measured by chemiluminescence using a Clarity™ Western ECL Substrate (Bio-Rad, Hercules, CA, USA) and ChemiDoc Imaging Systems (Bio-Rad).
2.8Statistical analysisThe data were analyzed using Student's t-test or one-way analysis of variance (ANOVA) for comparison between any two groups or among multiple groups, respectively. The program used was GraphPad Prism 6 (Northside Dr. Suite, San Diego, CA, USA). All data were expressed as mean±SEM and a p-value <0.05 was considered a significant difference.
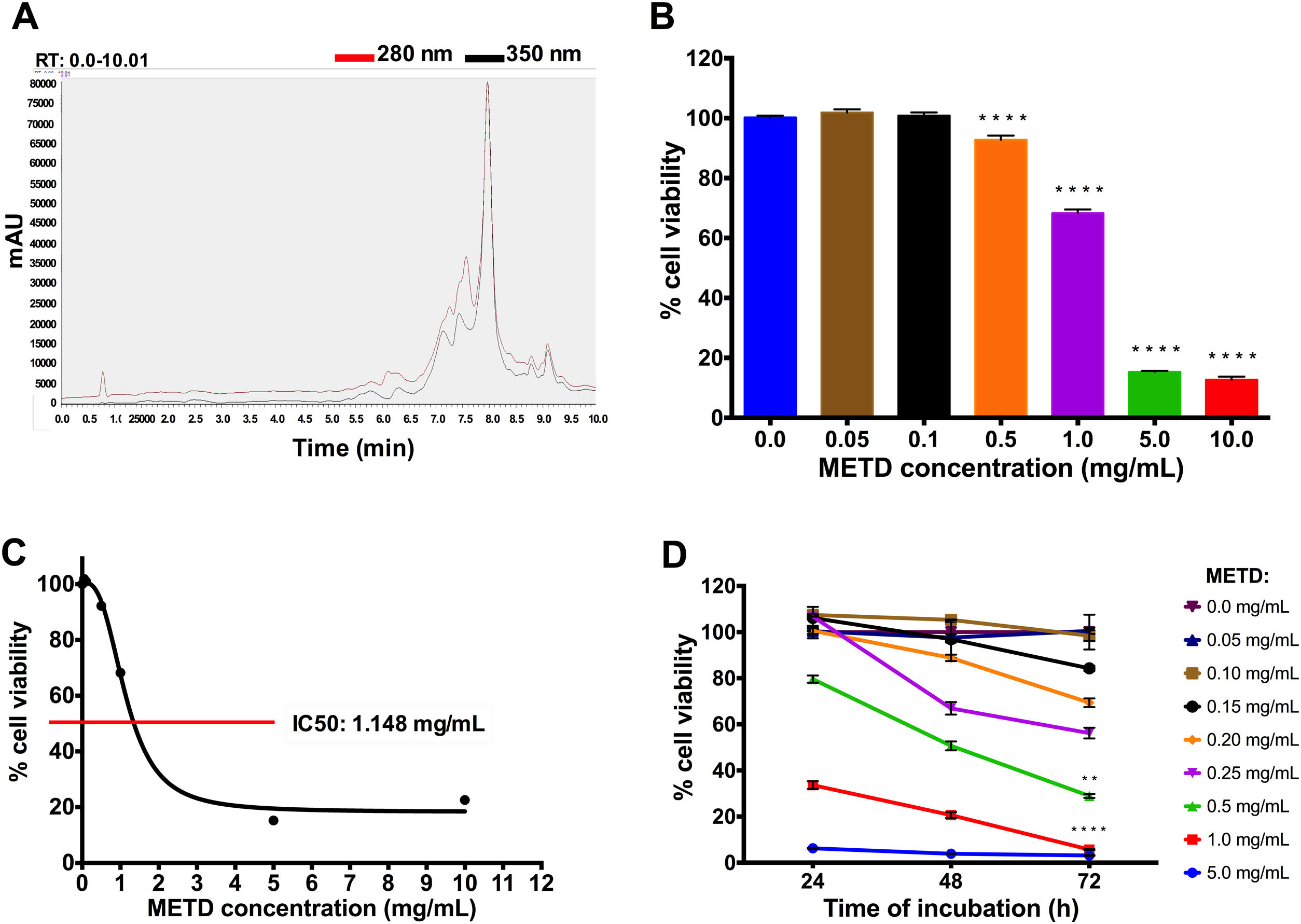
3Results3.1METD treatment is not cytotoxic for LX-2 cellsFirst, METD was analyzed by high-performance liquid chromatography with diode array detection (HPLC-DAD) for the quantification of hepatodamianol present in the METD. Fig. 1A shows a representation of a profile of METD measured (chromatogram) in milli-absorbance units (mAU) through time (retention time, RT). We identified that at a wavelength of 280 and 350nm, where a maximum RT can be observed at 8min, the hepatodamianol estimated concentration was between 5.35±0.12% [12].
 METD on the cell viability of LX-2. (A) Chromatographic profile of
METD on the cell viability of LX-2. (A) Chromatographic profile of Effect of METD on the cell viability of LX-2. (A) Chromatographic profile of METD by High-Performance Liquid Chromatography with Diode-Array Detection (HPLC-DAD). (B) Evaluation of cell viability at 24h. LX-2 cells were treated with the METD (0–10mg/mL) for 24h; viability was measured by the MTT assay, the bars indicate the percentage of cell viability. (C) IC50 of METD. D) Evaluation of cell viability at concentrations of 5.0 to 0.05mg/mL of METD for 24, 48, and 72h in LX-2 cells. All experiments were performed in triplicate. *p>0.05, **p>0.01, ***p>0.001 and ****p>0.0001.
Based on this, we then proceeded to determine the METD concentrations that are not cytotoxic for the LX-2 cell line by measuring cytotoxicity using an MTT assay. A range of METD concentrations from 0.05 to 10mg/mL at 24h (Fig. 1B) was used to treat cells up to 72h and choose the concentrations that do not decrease cell viability below 75%. We observed that cell viability was above 60% in concentrations less than 1mg/mL for 24h; contrary to that, a significant reduction in cell viability (<20%) was observed when cells were incubated at 5.0 and 10.0mg/mL (Fig. 1B). We also determined that METD 0.5mg/mL treatment at 24h, inhibited proliferation of LX-2 cells in a concentration-dependent manner (Fig. 1B). The inhibitory concentration 50 (IC50) of METD extract was 1.148mg/mL in LX-2 cells (Fig. 1C). Based on this, the evaluation of the cytotoxicity of the METD was performed using a range of concentrations from 0.05 to 5.0mg/mL in LX-2 cells at different time points (24, 48, and 72h). We found that METD 0.15mg/mL, decreases cell proliferation in a time-dependent manner (Fig. 1D). At concentrations of 1.0 and 5.0mg/mL METD inhibited proliferation at early time points (24h), while at concentrations of 200 to 500ng/mL cell viability decreased to less than 80% at 72h and concentrations less than 150ng/mL maintain cell viability around 80% (Fig. 1D). Based on these results, we decided to use the METD at the concentrations of 100 and 200ng/mL until 72h because it demonstrated no cytotoxic effect. This was done to further evaluate the effect of the METD in the presence of TGF-β in this cell line.
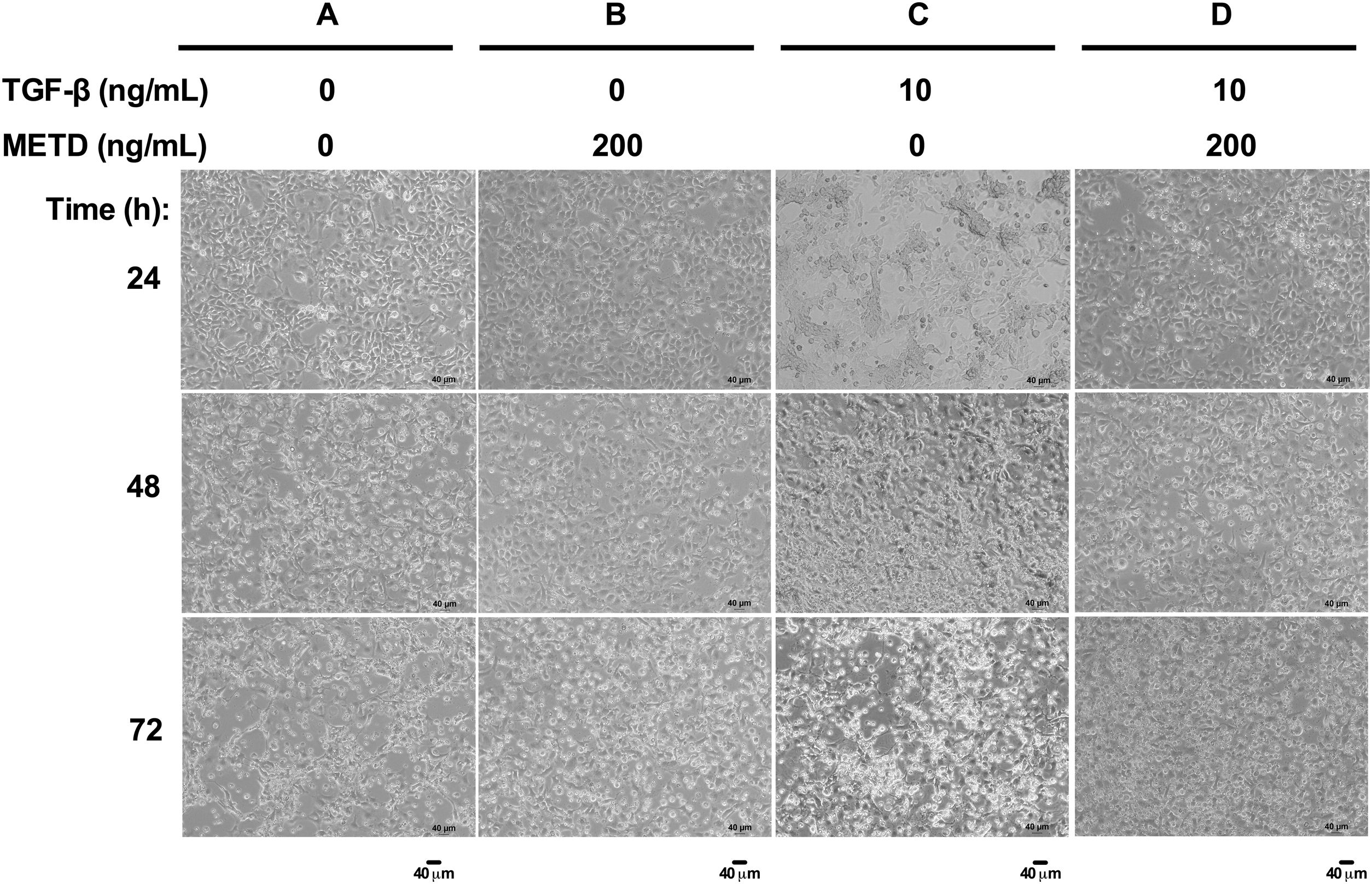
3.2METD does not modify sustained LX-2 cell morphology in the presence of TGF-β.To assess whether exposure to METD induces morphology disruption (Fig. 2), LX-2 cells were cultured in free-serum conditions in the presence of METD (200ng/mL) alone or combined with TGF-β (10ng/mL) at three different time points (24, 48 and 72h) and were evaluated under a phase-contrast microscope. We observed that LX-2 cells in the presence of METD (Fig. 2B, 24h) preserve a stellate shape, but when the cells were exposed to TGF-β (10ng/mL), cell morphology changed to an adherent spindle shape (stretching) forming clusters and leaving wide spaces between them (Fig. 2C, 24h). In addition, when the cells were exposed to combined treatment with TGF-β (10ng/mL) and METD (200ng/mL), this pattern was not observed (adherent spindle shape and clusters) and most of the cells kept their stellate shape (Fig. 2D, 24h). Most dead cells were observed upon treatment with TGF-β (10ng/mL) at 48h, and this effect was attenuated by the METD treatment. This pattern is similar to cells treated until 72h but showing a greater amount of cell death.
3.3METD attenuates fibrogenic expression markers in LX-2 cells. METD on the morphology of LX-2 cells treated with TGF-β. LX-2 cells were treated with 200ng/mL of
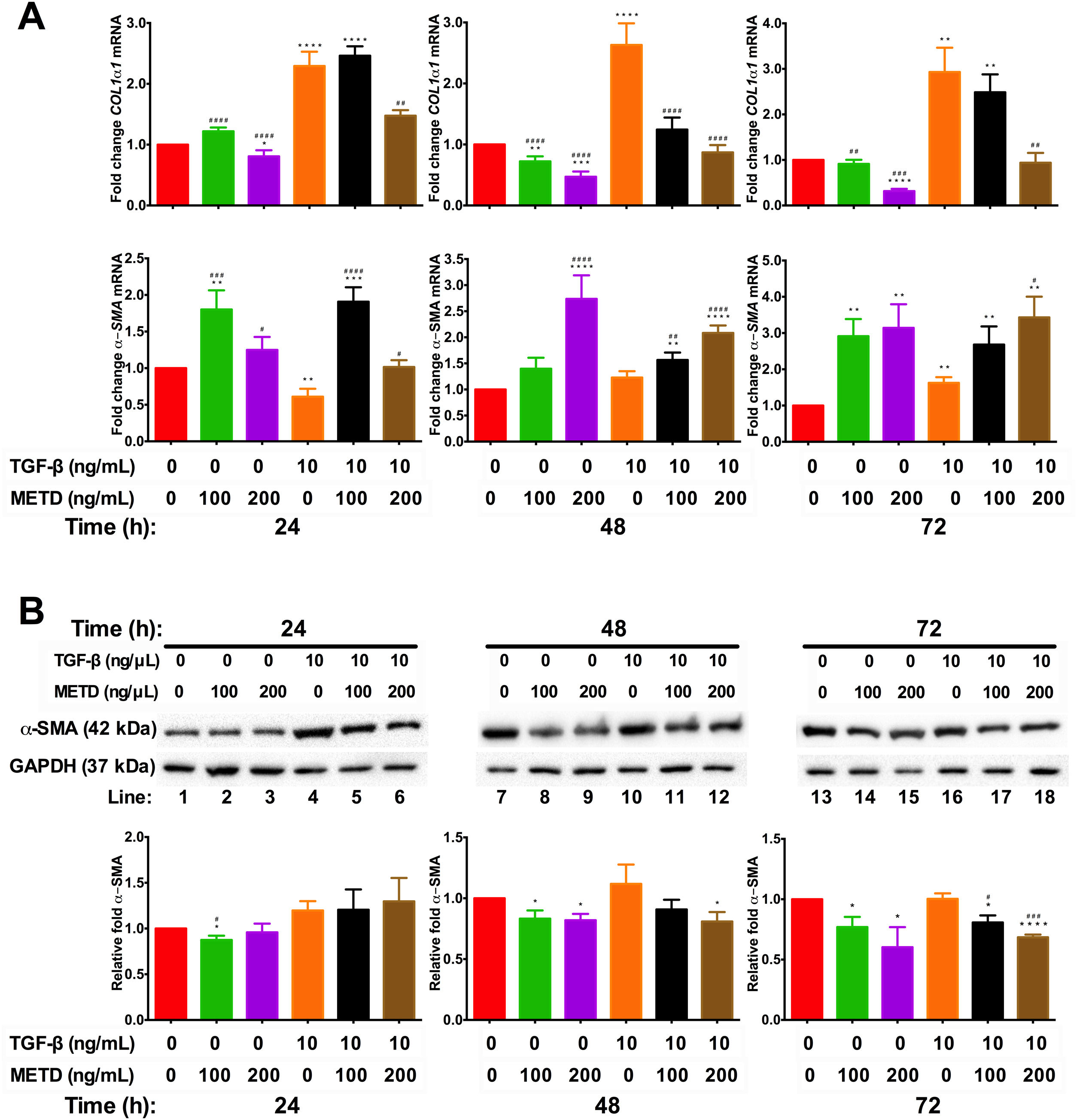
METD on the morphology of LX-2 cells treated with TGF-β. LX-2 cells were treated with 200ng/mL of We wanted to know whether METD could suppress LX-2 cell activation (Fig. 3). For this purpose, LX-2 cells were treated with METD (100 and 200ng/mL) alone o combined with TGF-β (10ng/mL) in FBS-free conditions at three different time points (24, 48, and 72h). We evaluated the expression of COL1α1-mRNA (Fig. 3A) through RT-qPCR in METD-treated cells at different times described above. We found that treatment with METD (100 or 200ng/mL) decreases COL1α1-mRNA levels expression. Only the concentration of 200ng/mL of METD inhibited COL1α1-mRNA expression at all times. We observed that, even at 72h, it has 65% more inhibition of COL1α1-mRNA expression than 100ng/mL of METD. As expected, treatment with TGF-β (10ng/mL) increased COL1α1-mRNA level expression at all times, and in the presence of METD, this effect was attenuated in both concentrations used (100 or 200ng/mL), but the concentration of 200ng/mL of METD presented the maximum effect in the inhibition of COL1α1-mRNA levels expression versus TGF-β (10ng/mL) (Fig. 3A), inhibiting expression at all times and being maintained until 72h, reaching the levels of basal COL1α1-mRNA expression from 48h to 72h (Fig. 3A).
 METD on pro-fibrotic markers in LX-2 cells treated with TGF-β. (A) COL1α1 and α-SMA mRNAs were measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by
METD on pro-fibrotic markers in LX-2 cells treated with TGF-β. (A) COL1α1 and α-SMA mRNAs were measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by Effect of METD on pro-fibrotic markers in LX-2 cells treated with TGF-β. (A) COL1α1 and α-SMA mRNAs were measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by METD treatment (100 or 200ng/mL) at 24, 48, and 72h, alone or combined with TGF-β (10ng/mL). β-actin mRNA expression was used as control. B) α-SMA protein expression was analyzed by Western blot. LX2 cells were plated in 6-well plates and treated as above. GAPDH protein was used as a control. All experiments were performed in triplicate. *Condition vs. SFB-free and #condition vs. TGF-β alone (*p<0.005, **p<0.01, ***p<0.001 and ****p<0.0001).
Furthermore, we evaluated transcriptional and translational α-SMA level expression (Fig. 3A and B, respectively). In the transcriptional evaluation, the presence of METD (100 or 200ng/mL) α-SMA-mRNA levels were increased and perpetuating up to 72h. Treatment with TGF-β (10ng/mL) increases α-SMA-mRNA level expression at 72h, and the presence of METD (100 or 200ng/mL) enhances this effect. METD (100ng/mL) decreases α-SMA protein expression at all times but at a concentration of 200ng/mL, this effect was observed only upon 48h (Fig. 3B). In the presence of TGF-β (10ng/mL) and METD, there was α-SMA protein downregulation from 48h at 200ng/mL of METD and was only observed at 72h for 100ng/mL of METD.
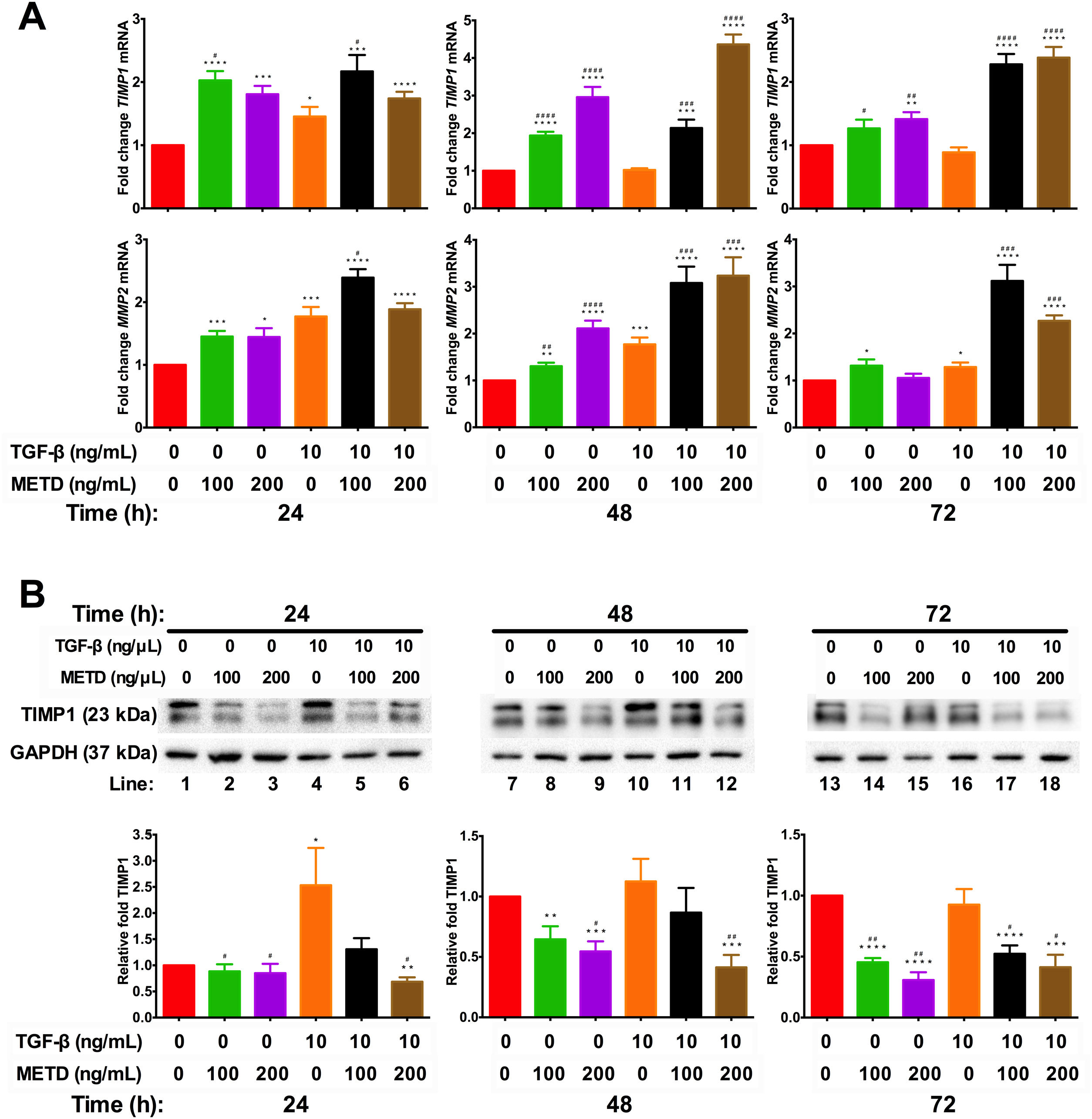
3.4METD modulates TIMP1 expressionSince we observed that METD lowers expression levels of COL1α1-mRNA and α-SMA protein, we wanted to assess whether METD was modifying extracellular matrix modulators such as MMP2 and TIMP1 (Fig. 4). Therefore, LX-2 cells were treated with METD (100 and 200ng/mL) alone o combined with TGF-β (10ng/mL) in FBS-free conditions at three different time points (24, 48, and 72h).
 METD on extracellular matrix modifiers in LX-2 cells treated with TGF-β. (A) TIMP1 and MMP2 mRNAs were measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by
METD on extracellular matrix modifiers in LX-2 cells treated with TGF-β. (A) TIMP1 and MMP2 mRNAs were measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by Effect of METD on extracellular matrix modifiers in LX-2 cells treated with TGF-β. (A) TIMP1 and MMP2 mRNAs were measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by METD treatment (100 or 200ng/mL) upon 24, 48, and 72h, alone or combined with TGF-β (10ng/mL). β-actin mRNA expression was used as control. (B) TIMP1 protein expression was analyzed by Western blot. LX2 cells were plated in 6-well plates and treated as above. GAPDH protein was used as a control. All experiments were performed in triplicate. *Condition vs. SFB-free and #condition vs. TGF-β alone (*p<0.005, **p<0.01, ***p<0.001 and ****p<0.0001).
We observed that MMP2-mRNA expression (Fig. 4A) was increased in cells in the presence of METD (100 and 200ng/mL) and when cells were further exposed to TGF-β (10ng/mL), this effect was enhanced. There was TIMP1-mRNA overexpression (Fig. 4A) in LX2 cells in presence of METD (100 and 200ng/mL) and the pattern repeated when the cells were also exposed to TGF-β (10ng/mL), showing enhanced overexpression levels. But TIMP1 protein expression (Fig. 4B) showed downregulation in LX2 cells in the presence of METD (48 and 72h), with 30% for 100ng/mL of METD to 44% for 200ng/mL of METD with greater inhibition at 72h; in combined treatment of TGF-β (10ng/mL) and METD, there was a decrease in TIMP1 protein expression levels at 72h (100ng/mL of METD) and at all times (200ng/mL of METD).
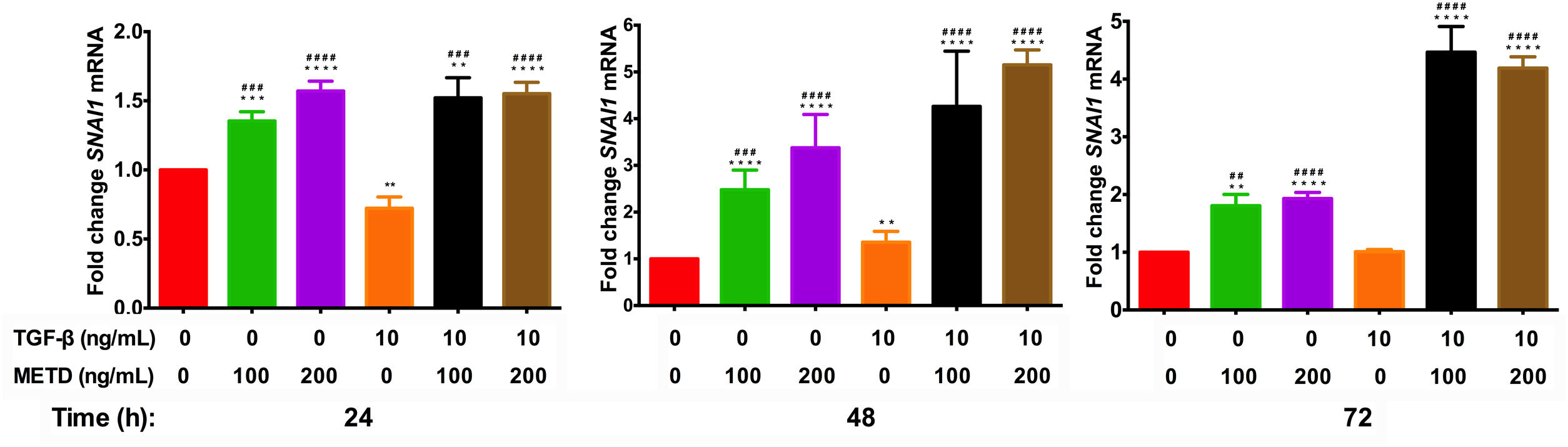
3.5METD increases SNAI1-mRNA expressionTo assess whether METD participates in the EMT mechanism (Fig. 5), LX-2 cells were treated with METD (100 and 200ng/mL) alone or combined with TGF-β (10ng/mL) in FBS-free conditions at three different time points (24, 48, and 72h) and then SNAI1-mRNA expression was evaluated. In the presence of METD, there was SNAI1-mRNA overexpression at all times, which increased even more than with stimulation with TGF-β (10ng/mL), and with the stimulation of both (METD and TGF-β), expression was up to 4 times more than the control.
 METD on SNAI1 mRNA expression in LX-2 cells treated with TGF-β.SNAI1 mRNA expression was measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by
METD on SNAI1 mRNA expression in LX-2 cells treated with TGF-β.SNAI1 mRNA expression was measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by Effect of METD on SNAI1 mRNA expression in LX-2 cells treated with TGF-β.SNAI1 mRNA expression was measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by METD treatment (100 or 200ng/mL) upon 24, 48, and 72h, alone or combined with TGF-β (10ng/mL). β-actin mRNA expression was used as a control. All experiments were performed in triplicate. *Condition vs. SFB-free and #condition vs. TGF-β alone (*p<0.005, **p<0.01, ***p<0.001 and ****p<0.0001).
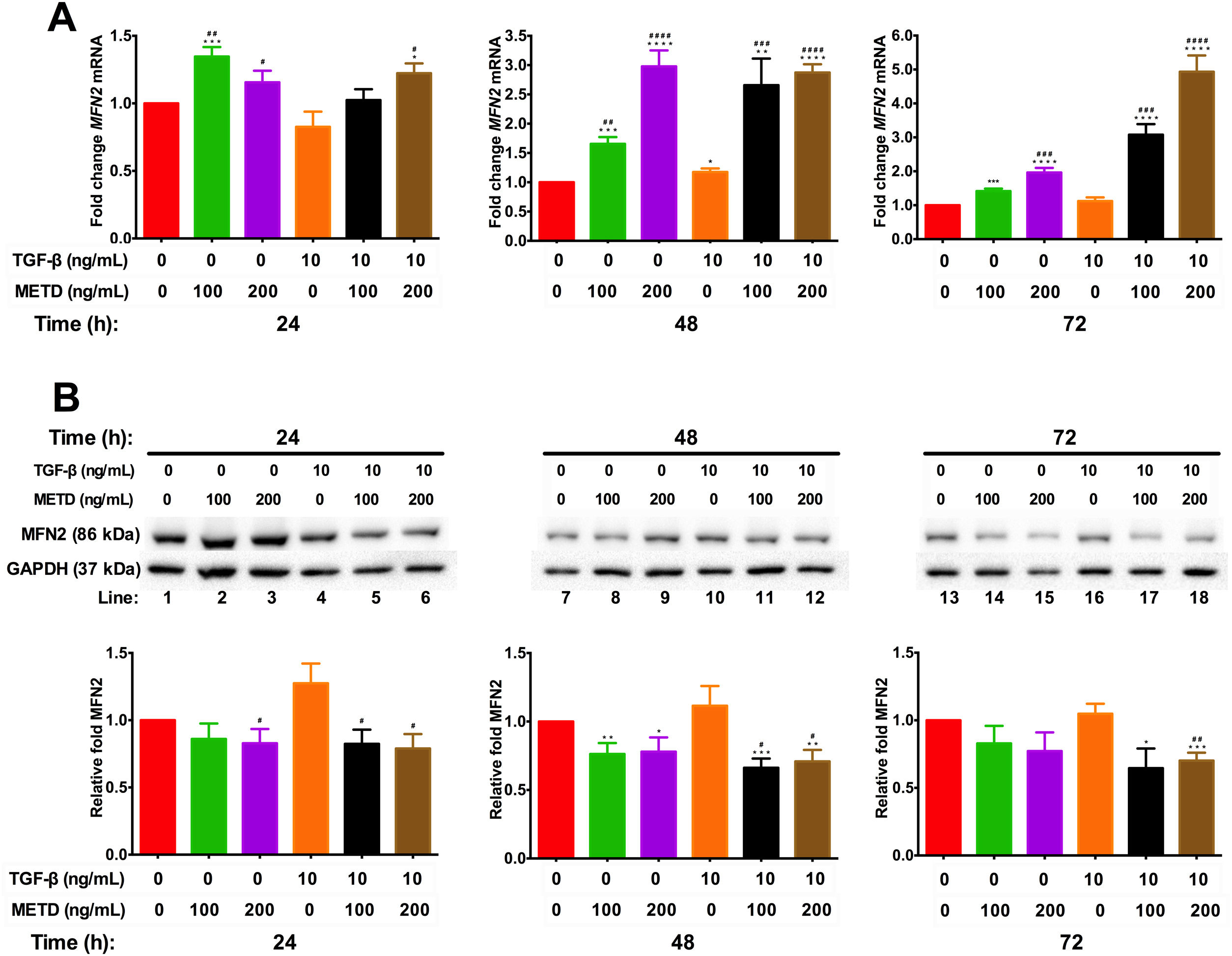
To assess mitochondrial function in the presence of METD, LX-2 cells were treated with METD (100 and 200ng/mL) alone or combined with TGF-β (10ng/mL) in FBS-free conditions at three different time points (24, 48, and 72h). We evaluated MFN2 mRNA and protein expression levels (Fig. 6) and found that MFN2-mRNA levels were upregulated in the presence of METD at all times, even more than upon stimulation with TGF-β. The stimulation with both (METD and TGF-β) also caused overexpression at all times and this effect was more evident in the dose of 200ng/mL of METD to 72h (Fig. 6A). MFN2 protein expression was decreased in the presence of METD at 24 (only for 200ng/mL) and 48h (both doses); in the presence of METD and TGF-β, it decreased compared to TGF-β at all times, even compared to control at 48 and 72h (Fig. 6B).
 METD on MFN2 mRNA and protein expression in LX-2cells treated with TGF-β. (A) MFN2 mRNAs were measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by
METD on MFN2 mRNA and protein expression in LX-2cells treated with TGF-β. (A) MFN2 mRNAs were measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by Effect of METD on MFN2 mRNA and protein expression in LX-2cells treated with TGF-β. (A) MFN2 mRNAs were measured by real-time PCR. LX2 cells were plated in 24-well plates, incubated in serum-free medium for 24h, followed by METD treatment (100 or 200ng/mL) upon 24, 48, and 72h, alone or combined with TGF-β (10ng/mL). β-actin mRNA expression was used as a control. (B) MFN2 protein expression was analyzed by Western blot. LX2 cells were plated in 6-well plates and treated as above. GAPDH was used as a control. All experiments were performed in triplicate. *Condition vs. SFB-free and #condition vs. TGF-β alone (*p<0.005, **p<0.01, ***p<0.001 and ****p<0.0001).
Hepatic stellate cells are a major cell type responsible for liver fibrosis following their activation into fibrogenic myofibroblast-like cells [14]. We explored the potential of METD as an antifibrotic agent and its role in EMT modulation and mitochondria dynamics in LX-2 cells. Liver fibrosis is in part the result of the imbalance between synthesis and degradation of ECM proteins. Activated HSCs express a combination of MMPs and TIMPs in different stages of the disease through the TGF-β pathway. They specifically express high levels of TIMP1 and MMP2. TIMP1 inhibits MMPs activity, perpetuating the accumulation of ECM whereas MMP2 activity is inhibited by TIMP1 [15,16].
COL1α1 is a marker used for fibrosis and METD causes a significant decrease in COL1α1-mRNA expression, 69% less than the basal expression of HSC in FBS-free conditions and the inhibition was dose-dependent. METD decreased COL1α1-mRNA expression in the presence of TGF-β also in a dose-dependent manner, even reaching the level of basal expression of HSC in FBS-free conditions, indicating that METD is a potent inhibitor of COL1α1-mRNA expression in a dose-dependent manner. In the fibrotic process, when the ECM is modified, in part by the accumulation of COL1α1 protein and cross-linking [17], it creates a pro-fibrotic environment that can activate MMP2 [18] and this, in turn, can activate TGF-β latent, and/or unlink the ECM integrins promoting apoptosis [19].
During HSC activation there is upregulation of smooth muscle-specific genes (among them α-SMA) and actin polymerization is increased. This promotes the release of ECM molecules like MRTF/A (actin-binding protein) and TGF-β activation that further increase the fibrotic response by stimulating the expression of genes such as α-SMA and COL1α1[20]. The METD increased α-SMA-mRNA expression and decreased α-SMA protein levels, in the presence or absence of TGF-β. This decrease of the α-SMA protein modulated by METD will diminish fibrotic stimulation and transmission of mechanical signals through actin polymerization, reducing the activation of TGF-β, release binding proteins to actin and therefore, lowering the fibrotic response by a decrease in α-SMA and COL1α1 expression.
We observed that LX-2 cells exposed to METD treatment alone or combined with TGF-β showed TIMP1-mRNA levels were upregulated compared with cells in FBS-free conditions but the METD in both concentrations used alone or combined with TGF-β decreased the levels of TIMP1 protein expression. Numerous studies suggest a positive association of TIMP1 with hepatic fibrogenesis and with progression to cancer [4]. TIMP1 inhibition is associated with a favorable fibrosis resolution but it has also been shown that the cell type and the microenvironment (a molecular network of factors) play a very important role; for example, the rigidity of the ECM [17] and the presence of other molecules [21,22].
According to the aforementioned, we expect that a decrease in TIMP1 protein expression could be modulated by METD and could have a favorable fibrotic resolution; however, the decrease or irruption of a stimulus would not be sufficient for the decrease or regression of a complex process, such as fibrosis [23].
MMPs are central to the fibrosis remodeling process and MMP activity is controlled at various levels: modulation of gene expression, compartmentalization, availability of activators, and inhibition [24,25]. METD alone or combined with TGF-β induced an increase in MMP2-mRNA expression with respect to cells in FBS-free conditions. MMP2 has a dose-dependent activation for COL1α1[24], if METD decreases the COL1α1 protein level, MMP2 activation could be affected. Also, MMP2 can activate latent TGF-β [26]; therefore, METD will decrease ECM deposition (a decrease in α-SMA and COL1α1 protein), and MMP2 and TGF-β activation. The induction of MMP2-mRNA overexpression by METD is not indicative of translational overexpression, so we do not know if METD is causing MMP2-mediated apoptosis [19].
SNAI1 is a downstream target of TGF-β and plays a key role in regulating several following cell fate decisions such as apoptosis and EMT induction (relates fibrogenesis to carcinogenesis) [5]. SNAI1-mRNA expression is different in distinct cell lines [27,28]. In this work, only at 48h, we found that SNAI1-mRNA levels were upregulated with the stimulation of TGF-β in relation to cells in FBS-free conditions. METD alone or combined with TGF-β increases SNAI1-mRNA expression. We hypothesized that SNAI1-mRNA overexpression is not reflected at the translational level or that there is some translational regulation that does not allow its activity because SNAI1 represses α-SMA-mRNA expression [29] and this does not happen, since, despite SNAI1-mRNA overexpression caused by the METD, there is α-SMA-mRNA overexpression at all times evaluated.
Mitochondria participate in almost all aspects of cell function and have been linked to pathogenic processes. Specifically, MFN2 expression has been related to chronic liver disease [8,9]. METD alone or combined with TGF-β causes MFN2-mRNA overexpression and decreased MFN2 protein expression. The modulation caused by METD and the probable cell triggering is not clear because MFN2 plays a role in the balance between apoptosis and autophagy and lack evaluation of the response caused in the cell context.
In this work, there were discrepancies in mRNA and protein expression in different genes (α-SMA, TIMP1, and MFN2) in the presence of METD alone or combined with TGF-β, indicating that METD is causing differential regulation of expression between mRNA and protein; thus, we hypothesize that transcriptional and translational regulatory mechanisms are being activated differently. These mechanisms of transcriptional regulation can be mediated by trans-acting factors, RNA-binding proteins, microRNA, and lncRNA that interact with cis-elements located in the mRNA, with activity pro-fibrotic or anti-fibrotic [30]. While translational regulation through protein degradation (ubiquitination) [30,31] could influence the discrepancy of mRNA and protein expression. Previous work [32] reported that miR-130 can regulate genes related to activation of HSC in a rat model, while miR-205 moderates the TGF-β/Smad signaling effect by direct downregulation of Smad [33].
We found evidence that METD in the presence of TGF-β decreases COL1α1-mRNA, α-SMA, and TIMP1 protein expression. It also decreases MFN2 and TIMP1 protein expression and overexpression of MMP2-mRNA. We suggested a probable mechanism by which METD could perform its hepatoprotective role is by inducing apoptosis of activated HSC cells. This effect must be verified in animal models, and the routes by which METD is modulating these markers should be explored. The increase in SNAI1-mRNA expression caused by METD was not explored in this work. Plant extracts, specifically flavonoids, are potential compounds in the search for antifibrotic drugs [34].
The fibrotic and HSC activation mechanism toward myofibroblast differentiation is a complicated molecular process in which multiple factors are involved; therefore, the mechanism for fibrogenesis reduction in vivo is complex and multifactorial. Cell type, the sequence of events, cell-cell interaction, and cell-ECM interaction intervene in the fibrotic process, in addition to the regulation triggered directly by a stimulus; therefore, the true activity of the molecules produced by the triggering of the stimulus will be subject to transcriptional, translational, functional and spatial regulation [17,30,31,35]. Further evidence should be generated to identify the related mechanisms involved in the hepatoprotective role of natural compounds.
Financial supportFinancial support was providing by PAICYT SA845-19 to Lozano-Sepulveda SA and CONACYT through the NATIONAL BIOBANK LABORATORY PROJECT: CONSOLIDATION 299077.
Conflict of interestThe authors have no conflicts of interest to declare.
Thanks to Dr. Natalia Martinez Acuña for her valuable comments.



