



Studies have shown that two polymorphisms were associated with steatosis and progression of non-alcoholic fatty liver disease (NAFLD) in different populations: the Patatin-like Phospholipase Domain Containing 3 (PNPLA3) and Transmembrane 6 Superfamily Member 2 (TM6SF2). However, the frequency and significance of these polymorphisms in an admixed population, i.e., Brazilian, is unknown. Therefore, we aimed to evaluate them in healthy subjects in comparison to patients with NAFLD.
Material and methodsThis was a multicenter cross-sectional study in 248 patients with biopsy-proven NAFLD and in 134 healthy controls from two tertiary centers in Brazil. PNPLA3 (rs738409 c.444C>G) and TM6SF2 (rs58542926 c.449C>T) polymorphisms were evaluated.
ResultsIn controls, the frequencies of PNPLA3 CC and CG+GG were 49.25% and 50.74%, respectively; in NAFLD patients, this was 31.05% and 68.88% (p=0.0044, 95% CI 1.037–2.977). PNPLA3 GG subjects had an increased risk (3.29-fold) of having NAFLD when compared to CC subjects (p=0.0044, 95% CI 1.504–7.225). In patients with nonalcoholic steatohepatitis (NASH), PNPLA3 GG compared to CC was associated with higher AST levels [38.4±25.3 versus 36.7±40.1IU/L, p=0.0395)] and with the presence of liver fibrosis (≥F2 fibrosis, p=0.0272). TM6SF2 polymorphisms were not in Hardy-Weinberg equilibrium in our NAFLD group precluding further analysis.
ConclusionWe demonstrated for the first time that PNPLA3 CG+GG increase the risk of NAFLD among Brazilian subjects. Moreover, PNPLA3 GG was associated with liver enzyme elevation and fibrosis in NASH patients.
Non-alcoholic fatty liver disease (NAFLD) is defined as hepatic accumulation of >5% fat in persons without significant alcohol consumption [1–3]. It represents a range of liver damage conditions from steatosis alone to the inflammatory phenotype—nonalcoholic steatohepatitis (NASH) [1]. The latter can progress to advanced fibrosis/cirrhosis with significant morbidity and mortality [4]. It is estimated that 24% of the world population has NAFLD with even higher proportions in South America (30.35%) [5,6]. However, it is interesting that only a small fraction of patients with NAFLD evolves to more advanced stages of the disease, revealing the interface between the environment and genetics in this complex polygenic illness [7]. Epidemiological, familial, and twin studies also provide evidence for genetic correlation with hepatic fat, NAFLD, and metabolic cirrhosis [8]. Moreover, the prevalence of NAFLD differs between geographic areas and ancestry groups as mentioned previously [6]. This also suggests a genetic susceptibility.
With the advent of genome wide association studies (GWAS), researchers have been able to partially unravel the pathophysiological mechanisms of several diseases including NAFLD [9]. In 2008, Romeo et al. identified single nucleotide polymorphisms (SNPs) in the Patatin-like Phospholipase Domain Containing 3 (PNPLA3) gene that is involved in the hepatocellular remodeling of lipid droplets and very low density lipoprotein (VLDL) secretion. This was shown to be one of the main determinants of inter individual differences and is related to the ethnical influence over the hepatic fat content [10]. These SNPs were highly associated with the hepatic accumulation of triacylglycerol as evaluated by magnetic resonance spectroscopy [10]. In 2014, Kozlitina et al. confirmed the association of PNPLA3 SNPs with hepatic steatosis and also found variants in the Transmembrane 6 Superfamily Member 2 (TM6SF2) gene associated with hepatic triglyceride content [11]. Since 2008, compelling data have linked PNPLA3 variants with risk for NAFLD and its severity [7,12–18]. TM6SF2 variants have also been implicated with these outcomes [19–21].
Despite the large amount of data from different parts of the world, the frequency and significance of these SNPs in Brazilian subjects is currently unknown [6]. Therefore, the aim of this study was to evaluate PNPLA3 and TM6SF2 SNPs in healthy Brazilian subjects in comparison to NAFLD patients and to evaluate its associations with hepatic steatosis and the severity of liver disease.
2Materials and methods2.1Clinical design and patients selectionThis was a multicenter cross-sectional study in healthy subjects and biopsy-proven NAFLD patients followed at the Outpatient Liver Unit of the Department of Gastroenterology at the University of Sao Paulo and at the Division of Gastroenterology (Gastrocentro) of the University of Campinas, Brazil. PNPLA3 (rs738409 c.444C>G) and TM6SF2 (rs58542926 c.449C>T) polymorphism were investigated in 248 patients with liver biopsy-proven NAFLD and 134 unrelated healthy volunteers. The comparison group consisted of non-obese and non-diabetic subjects without chronic hepatitis B, C or liver enzyme abnormalities. The inclusion criteria were age between 18 and 75 years old and biopsy-proven NAFLD. The exclusion criteria were any other chronic liver disease, significant alcohol intake (>100g/week), HIV infection, history of liver transplantation, previous exposure to drugs associated with fatty liver, or refusal to participate in the study.
NAFLD patient workup encompassed the exclusion of other liver diseases such as chronic hepatitis B and C, hemochromatosis, autoimmune hepatitis, Wilson's disease and alpha 1-antitrypsin deficiency. Liver histology was evaluated by an experienced liver pathologist, and specimens were classified according to the NASH Clinical Research Network [22]: steatosis (0–3), inflammation (0–3), and hepatocyte ballooning (0–2). The histological fibrosis scale varies from F0 to F4, and is classified as follows: F0: no fibrosis; F1: portal fibrosis without septa; F2: portal fibrosis with few septa; F3: fibrosis/bridge septa between the central and portal veins; and F4: cirrhosis [22].
2.2Variables evaluatedDemographic and anthropometric data were obtained (age, gender, weight, height, and body mass index) as well as comorbidities (diabetes mellitus, arterial hypertension, and dyslipidemia). Diagnosis of metabolic syndrome (MetS) was based on recommendations of the Adult Treatment Panel III Report if three or more of the following criteria were met: fasting glucose ≥110mg/dL, ≥130mmHg systolic or ≥85mmHg diastolic pressure, triglycerides ≥150mg/dL, high-density lipoproteins (HDL) <40mg/dL in men and <50mg/dL in women, and abdominal obesity [23]. Serum biochemistry included: fasting glucose, plasma insulin, total cholesterol and fractions, triglycerides, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma glutamyl transferase (GGT). These were collected after a 12-h overnight fast and evaluated at the time of the liver biopsy. The evaluation of insulin resistance relied on the Homeostatic Model of Assessment (HOMA-IR) calculation (22.5×fasting insulin [mU/mL]×glucose [mmol/L]). HOMA-IR ≥2.5 was used to define insulin resistance [24].
2.3DNA extraction and genotypingGenomic DNA was isolated from 200μL of blood by QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany). PNPLA3 (rs738409 c.444C>G) and TM6SF2 (rs58542926 c.449C>T) polymorphisms were genotyped using TaqMan primers and probes for allelic discrimination (7500 Fast Real-Time PCR system, Applied Biosystems, Thermo Fisher Brand, Foster City, CA, USA) per the manufacturer's recommendations. Direct genotyping was used in random samples to validate the results. Quality control was conducted to check the reproducibility of the results. The staff involved in genotyping was masked to the subject's clinical or biochemical data.
2.4Ethical considerationsThe Ethics Committees of the Clinics Hospitals of the University of Sao Paulo School of Medicine and of the University of Campinas approved this study (numbers 1,531,382 and 1,582,281, respectively). These protocols followed the 1975 Declaration of Helsinki. Informed consent was obtained from participants.
2.5Statistical analysisDescriptive statistics (means, standard deviations, minimum and maximum, and median values) were calculated. The chi-square test and Fisher's exact test were used to compare the categorical variables; Mann–Whitney and Kruskal–Wallis tests were used for numerical ones. For comparison of SNPs frequencies between groups—and for risk factor evaluation for NASH—logistic regression analysis was used and odds ratio (OR) were calculated in addition to a 95% confidence interval (CI). Hardy-Weinberg equilibrium was evaluated. Heterozygosity and polymorphic information content (PIC) in the studied groups were calculated [25]. A two-tailed probability value of <0.05 was considered to be significant. The SAS (Statistical Analysis System) for Windows, version 9.4 (SAS Institute Inc., 2002–2008, Cary, NC, USA) software package was used for the statistical analyses by biomedical statisticians from the Statistics Service at School of Medical Sciences of the University of Campinas.
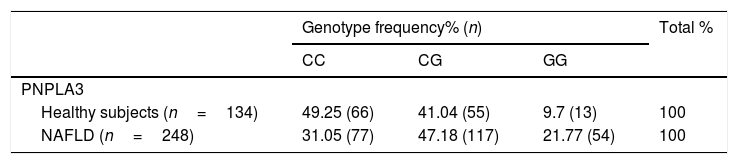
3ResultsWe included 248 patients with NAFLD (34 with steatosis and 214 with NASH) and compared them with 134 healthy subjects. The median age in the comparison group was 40.5 years old (19–78 years) and 56 years old in the NAFLD group (24–76 years); p<0.001. There were more male subjects in the comparison group than in the NAFLD group [65 (48.51%) versus 62 (25%), p<0.0001]. Since TM6SF2 polymorphisms in NAFLD group were not in Hardy-Weinberg equilibrium (chi-square test: 6.122, p=0.013), we excluded this polymorphism from the analyses. In the NAFLD group, the PNPLA3 heterozygosity was 0.6273 and the PIC was 0.5563. In the comparison group, the PNPLA3 heterozygosity was 0.5796, and the PIC was 0.4901. In healthy subjects, the frequencies of PNPLA3 CC and CG+GG were 49.25% and 50.74%, respectively; these were 31.05% and 68.88% in patients with NAFLD (p=0.0044, 95% CI 1.037–2.977; Table 1). After adjusting for age and gender on logistic regression analyses, PNPLA 3 GG subjects had a 3.29-fold higher risk of having NAFLD then CC subjects (p=0.0044, 95% CI 1.504–7.225). PNPLA3 CG subjects also had a 1.75-fold increased risk of having NAFLD versus CC individuals (p=0.0044, 95% CI 1.037–2.977; Table 1).
PNPLA3C>G polymorphisms frequencies in healthy subjects and patients with NAFLD.
| Genotype frequency% (n) | Total % | |||
|---|---|---|---|---|
| CC | CG | GG | ||
| PNPLA3 | ||||
| Healthy subjects (n=134) | 49.25 (66) | 41.04 (55) | 9.7 (13) | 100 |
| NAFLD (n=248) | 31.05 (77) | 47.18 (117) | 21.77 (54) | 100 |
Genotype frequency and logistic regression analysis. Abbreviations: NAFLD: non-alcoholic fatty liver disease; OR: Odds Ratio. p=0.0044 (CG×CC: OR 1.757, 95% CI 1.037–2.977; GG×CC: OR 3.296, 95% CI 1.504–7.225).
Likewise, the presence of the G allele of PNPLA3 was associated with NAFLD (p=0.0044, OR 2.054, 95% CI 1.251–3.371) in relation to the comparison group. When separately evaluating the G allele in steatosis patients and NASH patients in relation to subjects without NAFLD, the presence of the risk allele was associated with a 2.29-fold increased risk of having steatosis (p=0.0052, 95% CI 1.367–3.838). No significant differences were observed between NASH and control subjects (OR 1.237, 95% CI 0.567–2.699) or NASH versus steatosis (OR 1.852, 95% CI 0.880–3.896).
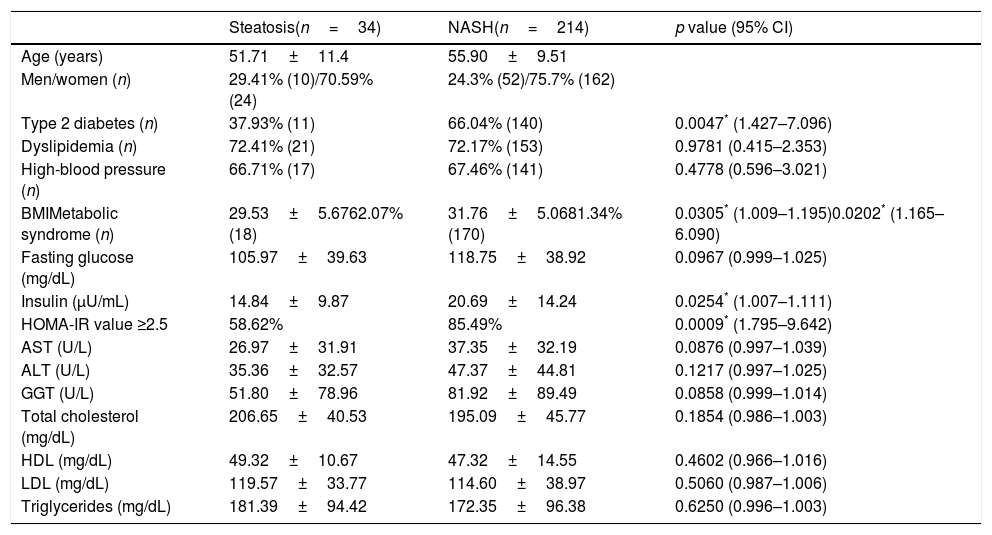
The anthropometric, clinical and biochemical characteristics of patients with NAFLD (n=248) are provided in Table 2. Those with NASH (n=214) were older, had a higher frequency of diabetes (p=0.0047, 95% CI 1.427–7.096), metabolic syndrome (p=0.0202, 95% CI 1.165–6.090), insulin resistance (p=0.0009, 95% CI 1.795–9.642), and had a higher BMI (p=0.0305, 95% CI 1.009–1.195) than patients with steatosis (n=34). On a stepwise multivariate logistic regression analyses, diabetes was associated with 3.32-fold increased risk of NASH among patients with NAFLD (p=0.0320, OR 3.32, 95% CI 1.148–9.944), and obesity was associated with 10.597-fold increased risk of NASH in the same population (p=0.0038, OR 10.587, 95% CI 2.594–43.286).
Demographic, clinical and biochemical characteristics of patients with NAFLD.
| Steatosis(n=34) | NASH(n=214) | p value (95% CI) | |
|---|---|---|---|
| Age (years) | 51.71±11.4 | 55.90±9.51 | |
| Men/women (n) | 29.41% (10)/70.59% (24) | 24.3% (52)/75.7% (162) | |
| Type 2 diabetes (n) | 37.93% (11) | 66.04% (140) | 0.0047* (1.427–7.096) |
| Dyslipidemia (n) | 72.41% (21) | 72.17% (153) | 0.9781 (0.415–2.353) |
| High-blood pressure (n) | 66.71% (17) | 67.46% (141) | 0.4778 (0.596–3.021) |
| BMIMetabolic syndrome (n) | 29.53±5.6762.07% (18) | 31.76±5.0681.34% (170) | 0.0305* (1.009–1.195)0.0202* (1.165–6.090) |
| Fasting glucose (mg/dL) | 105.97±39.63 | 118.75±38.92 | 0.0967 (0.999–1.025) |
| Insulin (μU/mL) | 14.84±9.87 | 20.69±14.24 | 0.0254* (1.007–1.111) |
| HOMA-IR value ≥2.5 | 58.62% | 85.49% | 0.0009* (1.795–9.642) |
| AST (U/L) | 26.97±31.91 | 37.35±32.19 | 0.0876 (0.997–1.039) |
| ALT (U/L) | 35.36±32.57 | 47.37±44.81 | 0.1217 (0.997–1.025) |
| GGT (U/L) | 51.80±78.96 | 81.92±89.49 | 0.0858 (0.999–1.014) |
| Total cholesterol (mg/dL) | 206.65±40.53 | 195.09±45.77 | 0.1854 (0.986–1.003) |
| HDL (mg/dL) | 49.32±10.67 | 47.32±14.55 | 0.4602 (0.966–1.016) |
| LDL (mg/dL) | 119.57±33.77 | 114.60±38.97 | 0.5060 (0.987–1.006) |
| Triglycerides (mg/dL) | 181.39±94.42 | 172.35±96.38 | 0.6250 (0.996–1.003) |
Univariate logistic regression analysis. Abbreviations: CI: confidence interval; ALT: alanine aminotransferase; AST: aspartate aminotransferase; BMI: body mass index; HDL: high-density lipoprotein; HOMA-IR: Homeostatic Model of Assessment; LDL: low-density lipoprotein; GGT: gamma glutamyl transferase; NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis.
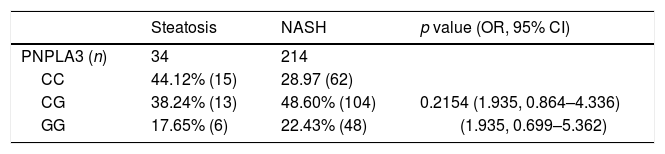
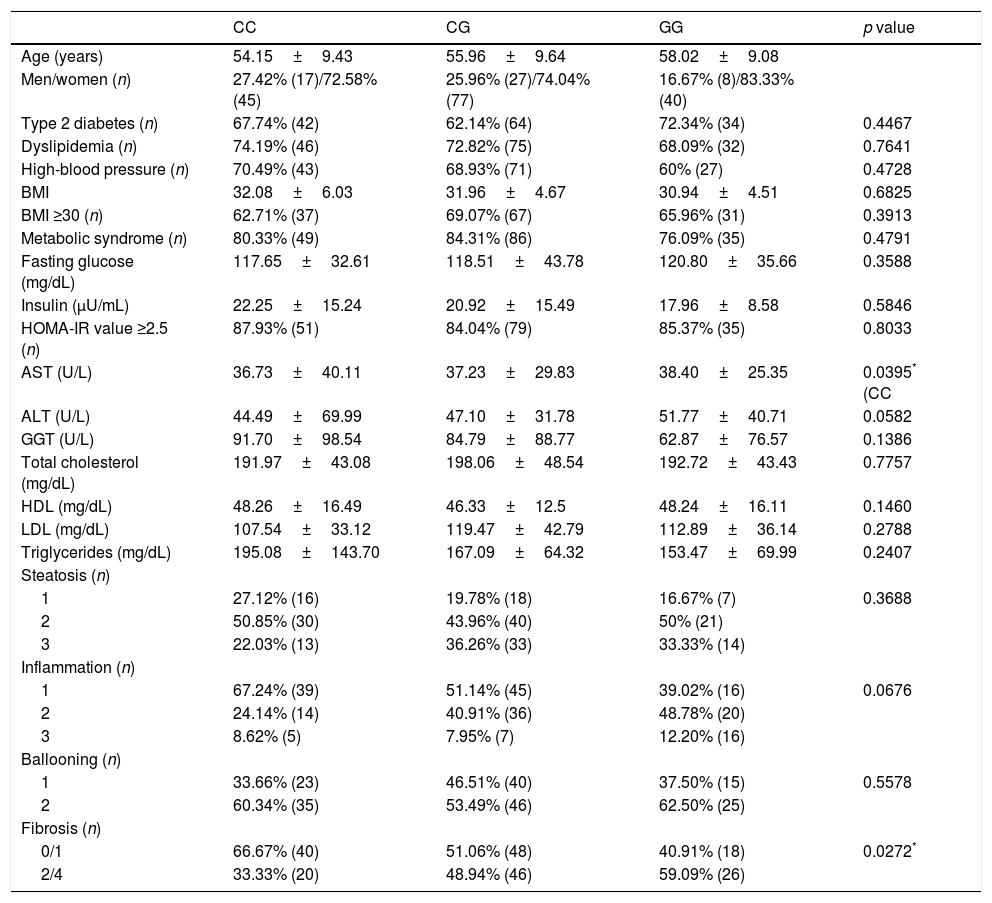
PNPLA3 genotypes, however, were unable to predict NASH among patients with NAFLD (p=0.2154) as shown in Table 3. Similarly, the presence of the G allele of PNPLA3 was not a risk factor of NASH among NAFLD patients (p=0.0797, OR 1.935, 95% IC 0.925–4.051). In NASH patients, PNPLA3 GG was associated with higher AST levels [38.4±25.3U/L versus 36.7±40.1IU/L, p=0.0395)] when compared to CC. It was also associated with significant liver fibrosis (≥Stage 2 fibrosis), p=0.0272 (Table 4).
PNPLA3C>G polymorphisms frequencies in patients with NAFLD.
| Steatosis | NASH | p value (OR, 95% CI) | |
|---|---|---|---|
| PNPLA3 (n) | 34 | 214 | |
| CC | 44.12% (15) | 28.97 (62) | |
| CG | 38.24% (13) | 48.60% (104) | 0.2154 (1.935, 0.864–4.336) |
| GG | 17.65% (6) | 22.43% (48) | (1.935, 0.699–5.362) |
Genotype frequency and logistic regression analysis. Abbreviations: NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; OR: odds ratio; CI: confidence interval.
Associations of PNPLA3 polymorphisms in patients with NASH (n=214).
| CC | CG | GG | p value | |
|---|---|---|---|---|
| Age (years) | 54.15±9.43 | 55.96±9.64 | 58.02±9.08 | |
| Men/women (n) | 27.42% (17)/72.58% (45) | 25.96% (27)/74.04% (77) | 16.67% (8)/83.33% (40) | |
| Type 2 diabetes (n) | 67.74% (42) | 62.14% (64) | 72.34% (34) | 0.4467 |
| Dyslipidemia (n) | 74.19% (46) | 72.82% (75) | 68.09% (32) | 0.7641 |
| High-blood pressure (n) | 70.49% (43) | 68.93% (71) | 60% (27) | 0.4728 |
| BMI | 32.08±6.03 | 31.96±4.67 | 30.94±4.51 | 0.6825 |
| BMI ≥30 (n) | 62.71% (37) | 69.07% (67) | 65.96% (31) | 0.3913 |
| Metabolic syndrome (n) | 80.33% (49) | 84.31% (86) | 76.09% (35) | 0.4791 |
| Fasting glucose (mg/dL) | 117.65±32.61 | 118.51±43.78 | 120.80±35.66 | 0.3588 |
| Insulin (μU/mL) | 22.25±15.24 | 20.92±15.49 | 17.96±8.58 | 0.5846 |
| HOMA-IR value ≥2.5 (n) | 87.93% (51) | 84.04% (79) | 85.37% (35) | 0.8033 |
| AST (U/L) | 36.73±40.11 | 37.23±29.83 | 38.40±25.35 | 0.0395* (CC |
| ALT (U/L) | 44.49±69.99 | 47.10±31.78 | 51.77±40.71 | 0.0582 |
| GGT (U/L) | 91.70±98.54 | 84.79±88.77 | 62.87±76.57 | 0.1386 |
| Total cholesterol (mg/dL) | 191.97±43.08 | 198.06±48.54 | 192.72±43.43 | 0.7757 |
| HDL (mg/dL) | 48.26±16.49 | 46.33±12.5 | 48.24±16.11 | 0.1460 |
| LDL (mg/dL) | 107.54±33.12 | 119.47±42.79 | 112.89±36.14 | 0.2788 |
| Triglycerides (mg/dL) | 195.08±143.70 | 167.09±64.32 | 153.47±69.99 | 0.2407 |
| Steatosis (n) | ||||
| 1 | 27.12% (16) | 19.78% (18) | 16.67% (7) | 0.3688 |
| 2 | 50.85% (30) | 43.96% (40) | 50% (21) | |
| 3 | 22.03% (13) | 36.26% (33) | 33.33% (14) | |
| Inflammation (n) | ||||
| 1 | 67.24% (39) | 51.14% (45) | 39.02% (16) | 0.0676 |
| 2 | 24.14% (14) | 40.91% (36) | 48.78% (20) | |
| 3 | 8.62% (5) | 7.95% (7) | 12.20% (16) | |
| Ballooning (n) | ||||
| 1 | 33.66% (23) | 46.51% (40) | 37.50% (15) | 0.5578 |
| 2 | 60.34% (35) | 53.49% (46) | 62.50% (25) | |
| Fibrosis (n) | ||||
| 0/1 | 66.67% (40) | 51.06% (48) | 40.91% (18) | 0.0272* |
| 2/4 | 33.33% (20) | 48.94% (46) | 59.09% (26) | |
Chi-square test, Fisher's exact test and Kruskal–Wallis test. Abbreviations: ALT: alanine aminotransferase; AST: aspartate aminotransferase; BMI: body mass index; HDL: high-density lipoprotein; HOMA-IR: Homeostatic Model of Assessment; LDL: low-density lipoprotein; GGT: gamma glutamyl transferase; MetS: metabolic syndrome; NASH: nonalcoholic steatohepatitis.
This is the first study in Brazilian subjects with and without NAFLD evaluating the roles of PNPLA3 and TM6SF2 polymorphisms over fatty liver occurrence and its severity. In this cross-sectional clinical multicenter study, the G allele of PNPLA3 was associated with an increased risk of having NAFLD, after adjusting for age and gender. In addition, the PNPLA3 genotype demonstrated a dose effect with the heterozygote (CG). There was an intermediate risk between CC and GG homozygotes over the occurrence of NAFLD (odds ratio of 1.757 for CG and of 3.296 for GG compared to the CC genotype). When addressing PNPLA3 SNPs in patients with NASH, the GG genotype was associated with higher AST levels and to a higher frequency of significant liver fibrosis (≥Stage 2 fibrosis) compared to the CC genotype. TM6SF2 polymorphisms were not in Hardy-Weinberg equilibrium in our NAFLD group precluding further analysis.
The PNPLA3 gene encodes a membrane protein located in the endoplasmic reticulum on the surface of the lipid droplets. This protein is highly expressed in human hepatocytes and hepatic stellate cells (HSCs) and has hydrolase activity [14]. The substitution of isoleucine by methionine at position 148 of the PNPLA gene leads to the production of a loss of function protein (I148M) rather than a functional enzyme directed to triglycerides and retinyl ester degradation in hepatocytes and HSCs, respectively [26–28]. The end result is the retention of mutated protein on the lipid droplets evading proteasomal degradation and impairment of fatty acid mobilization from hepatocytes [29]. The fact that the abnormal protein remains retained in the fat droplet may hamper access of other lipases to the droplet itself and may culminate in the NAFLD phenotype [30].
Indeed, two recent meta-analysis have linked the PNPLA3 G allele with an increased risk of fatty liver disease [31,32]: The odds ratios for NAFLD ranges from 3.31 to 3.41 when the GG PNPLA3 genotype was present, similar to our results. In a recent Anatolian population based multicenter study evaluating 216 patients with biopsy-proven NAFLD and 150 controls, Uygun et al. found that the PNPLA3 rs738409 GG polymorphism was associated with a 27-fold increased risk of development of NAFLD and also an association with the inflammatory severity of liver injury regardless of clinical and metabolic risk factors [33].
In contrast to this study, we found that the high risk phenotype conferred by PNPLA3 was associated with hepatic fibrosis, confirming the results obtained in other studies evaluating several populations [12–14,34,35]. The current understanding of the mechanistic effects of PNPLA3 variants over liver fibrosis came from recent experimental studies. The I148M mutation leads to retinol retention in HSC [14]. Indeed, Kovarova et al. demonstrated that liver extracts from PNPLA3 GG genotype subjects had an increased concentration of retinyl-palmitate, retinyl linoleate, and retinyloleate [36]. Retinol retention in the liver could translate clinically to a lower fasting circulating retinol concentration versus those carrying the PNPLA3 148M allele [37]. However, there is release of stored retinoids during HSC activation toward a myofibroblast-like phenotype in hepatic fibrogenesis—this might seem paradoxical. Interestingly, Pingitore et al. evaluated human HSCs and suggested that PNPLA3-mediated retinol release in the absence of a loss of function variant might actually be a protective mechanism against fibrosis [38]. The authors suggest that once the HSC is activated, an upregulation of the wild type PNPLA3 protein occurs obviating fibrosis through downregulation of matrix metalloproteinase (MMP) 2, tissue inhibitor of metalloproteinase (TIMP) 1, and TIMP2—mechanism mediated by retinoids. The I148M mutation carriers therefore have retinoid retention and a loss of feedback mechanism against fibrosis [38].
Bruschi et al. have also studied PNPLA3-associated fibrosis processes [39]. Primary human HSCs containing the I148M variant showed a more inflammatory and fibrogenic phenotype. Pro-inflammatory cytokines identified as involved were the chemokine (C–C motif) ligand (CCL) 5 and granulocyte-macrophage colony-stimulating factor (GM-CSF). These were related to immune cell migration. Moreover, I148M HSCs have an unbalanced fatty acid composition and increased lipid droplets that up-regulate c-Jun N-terminal kinase (JNK) signaling. The final outcome of these events was a proinflammatory status and fibrosis—the latter is through peroxisome proliferator-activated receptor gamma (PPARγ) promoter binding/activity decrease [39].
The TM6SF2 gene encoded protein is involved in the intracellular transport and lipidation of VLDL and its secretion from hepatocytes [34,40,41]. The substitution of glutamic acid by lysine at protein residue 167 (E167K) is associated with a reduction in the protein expression in Huh7 cells and in the liver. This leads to liver triglyceride accumulation and lower circulating lipoproteins [34,42]. Interestingly, TM6SF2 SNP loss of function leads to triglyceride retention in the liver and decreases the likelihood of hypertriglyceridemia and cardiovascular events [11,19,20]. The minor T allele of TM6SF2 [Lys167(K)] has been linked to steatosis, advanced fibrosis, and cirrhosis in several studies [11,19–21,42–44].
Data from our NAFLD patients confirmed the impact of well known risk factors for steatohepatitis: older age, diabetes, obesity, and metabolic syndrome [5,45]. PNPLA3 SNPs were unable to predict NASH in these patients however. The inability to discriminate NASH within the NAFLD population could be attributed, at least in part, to the small number of patients with steatosis (n=34) enrolled in our study. The cohort is derived from tertiary health centers, and referral bias could explain the high proportion of NASH versus steatosis only. A study of Brazilian subjects from primary or secondary care could lead to different proportions and conclusions.
There is a lack of data on PNPLA3 and TM6SF2 SNPs in healthy subjects in South America [6]. Sookoian et al. studied an Argentine cohort and found a PNPLA3 G allele frequency of 33.6%, and TM6SF2 T allele frequency of only 5.5% [44,46]. Also in Argentina, Pontoriero et al. described PNPLA3 GG genotype as present in 55.4% of healthy male volunteers [47]. This is a much higher frequency than in our population (9.7%). In contrast to Argentina, Brazil has a higher mixture of ethnicities whose genetic background is mainly European, African, and Native Brazilian Amerindians [48]. Our previous report confirmed the reflex of this admixed country population in our patients with NASH (genetic ancestry contribution: European 48.8%, African 41.7%, and Amerindian 9.5%) [49]. It is important to emphasize that even in this genetic diversity scenario, the PNPLA3 I148M variant was demonstrated to be an important risk factor for NAFLD and is associated with significant liver fibrosis in NASH as observed in other populations around the world.
A recent systematic review of PNPLA3 polymorphism in different regions and among different ethnic groups (2504 subjects) showed that the CC, CG, and GG genotypes frequencies were 56.9%, 33.8%, and 9.3%, respectively [13]. These findings are quite similar to ours (49.25%, 41.04%, and 9.7%, respectively). While not currently recommended for routine clinical care [1–3], PNPLA3 genotyping could be an important tool to stratify risk of NAFLD and progressive fatty liver disease across different regions. Toward this reality, increasing efforts have recently been undertaken to evaluate the role of genotyping incorporation for identification, assessment, and risk stratification of patients with fatty liver disease [50–52].
In conclusion, we demonstrate for the first time that PNPLA3 CG+GG increases the risk of NAFLD among Brazilian subjects. Moreover, PNPLA3 GG is associated with AST elevation and fibrosis in patients with biopsy-proven NAFLD as observed in other populations. Further studies in Brazilian NAFLD patients with a larger healthy comparison group are recommended to validate these findings.AbbreviationsAST
aspartate aminotransferase
ALTalanine aminotransferase
CCLchemokine (C–C motif) ligand
CIconfidence interval
GGTgamma glutamyl transferase
GM-CSFgranulocyte-macrophage colony-stimulating factor
GWASgenome wide association studies
HDLhigh-density lipoproteins
HSCshepatic stellate cells
HOMA-IRHomeostatic Model of Assessment
JNKc-Jun N-terminal kinase
MetSmetabolic syndrome
MMPmatrix metalloproteinase
NAFLDnon-alcoholic fatty liver disease
NASHnonalcoholic steatohepatitis
ORodds ratios
PNPLA3Patatin-like Phospholipase Domain Containing 3
PPARγperoxisome proliferator-activated receptor gamma
SNPssingle nucleotide polymorphisms
TIMPtissue inhibitor of metalloproteinase
TM6SF2Transmembrane 6 Superfamily Member 2
Ethical considerationsThe Ethics Committees of the Clinics Hospitals of the University of Sao Paulo School of Medicine and of the University of Campinas approved this study (numbers 1,531,382 and 1,582,281, respectively). These protocols followed the 1975 Declaration of Helsinki. Informed consent was obtained from participants.
FundingNone to declare.
Author contributionsMazo DF and Oliveira CP conceived and designed the study, contributed to the data analysis and interpretation and wrote the manuscript. Malta FM, Salles APM and Gomes-Gouvea MS performed the polymorphism genotyping. Mazo DF, Stefano JT, and Nastri ACS collected and assembled the data. Almeida JR, Pinho JRR, Carrilho FJ, and Oliveira CP reviewed the manuscript critically. All authors read and approved the final version of the manuscript. Mazo DF is responsible for the integrity of the work as a whole.
Conflict of interestThe authors report no conflicts of interest in this work.



