



La miocardiopatía/displasia arritmogénica del ventrículo derecho es una enfermedad hereditaria autosómica dominante con una prevalencia estimada de 1:2,500–1:5,000, siendo mayor en el género masculino (3:1). Se caracteriza histológicamente por reemplazo de los cardiomiocitos por tejido fibroadiposo, lo cual predispone a arritmias ventriculares, insuficiencia ventricular derecha y muerte súbita cardiaca. El objetivo principal del tratamiento es reducir el riesgo de muerte súbita y mejorar la calidad de vida de los pacientes. Se presenta el caso de una mujer de 23 años con clínica de palpitaciones, dolor torácico con actividad física, síncope y cefalea iniciada hace 6 años durante un primer embarazo. Por aumento de sintomatología se realizó prueba de esfuerzo, durante la cual presentó taquicardia ventricular monomórfica sostenida colapsante. Se realizó cardiorresonancia, evidenciando dilatación ventricular derecha, aumento en su trabeculación y disminución de su función. Se realizó ablación con mapeo tridimensional; durante la comprobación con infusión de isoproterenol se generó flutter ventricular polimórfico requiriendo cardioversión eléctrica. Se decidió implantar cardiodesfibrilador bicameral y realizar ablación de ganglio estrellado como prevención secundaria. Tras su egreso reconsultó múltiples veces por descargas del dispositivo asociadas a palpitaciones. Se realizó una revisión exhaustiva de la historia clínica encontrando que la paciente tiene características sugestivas de displasia arritmogénica del ventrículo derecho por lo cual se aplican los criterios de Task Force concluyendo que, al cumplir con más de 2 criterios mayores, la paciente presentaba un diagnóstico definitivo de esta enfermedad.
Arrhythmogenic right ventricular cardiomyopathy/dysplasia is an inherited autosomal dominant disease, with an estimated prevalence of 1:2,500 to 1:5,000, being higher in males (3:1). It is characterised histologically by the substitution of cardiomyocytes for fibrous-adipose tissue, which predisposes to ventricular arrhythmias, right ventricular failure, and sudden cardiac death. The main aim of treatment is to reduce the risk of sudden death and improve the quality of life of patients. The case is presented of a 23 year old woman whose clinical symptoms started with palpitations, chest pain with physical activity, syncope, and headache, 6 years ago during her first pregnancy. Due to an increase in symptomatology, a stress test was performed, during which she collapsed with a sustained monomorphic ventricular tachycardia. A cardiac magnetic resonance scan showed dilation, an increase in trabeculae, and decreased function of the right ventricle. A 3-dimensional mapping and ablation was performed, and during the isoproterenol infusion test, a polymorphic ventricular flutter was generated that required electrical cardioversion. The decision was made to implant a dual chamber cardioverter defibrillator and perform stellate ganglion ablation as secondary prevention. After her discharge, the patient re-consulted many times due to discharges of the device associated with palpitations. A comprehensive review of the patient's medical records was performed, finding characteristics that may suggest arrhythmogenic right ventricular dysplasia. The Task Force criteria was applied, concluding that, as she met more than 2 major criteria, the patient had a definitive diagnosis of this disease.
La miocardiopatía/displasia arritmogénica del ventrículo derecho (M/DAVD) es una enfermedad hereditaria autosómica dominante caracterizada histológicamente por el reemplazo de los cardiomiocitos por tejido fibroadiposo; lo cual predispone a arritmias ventriculares, insuficiencia ventricular derecha, y muerte súbita cardiaca (MSC)1–3. Desde su primera descripción, se han realizado esfuerzos por mejorar su comprensión, planteando nuevas estrategias centradas en un diagnóstico precoz mediante la evaluación de las alteraciones cardiacas funcionales y estructurales características de esta entidad, así como la realización de pruebas genéticas en los pacientes considerados de alto riesgo. El objetivo principal del tratamiento es reducir el riesgo de MSC y mejorar la calidad de vida de los pacientes2,4,5. Este artículo tiene como objetivo reportar el caso de una paciente con síncope quien después de varios estudios complementarios es diagnosticada con M/DAVD, así como realizar la revisión bibliográfica del tema.
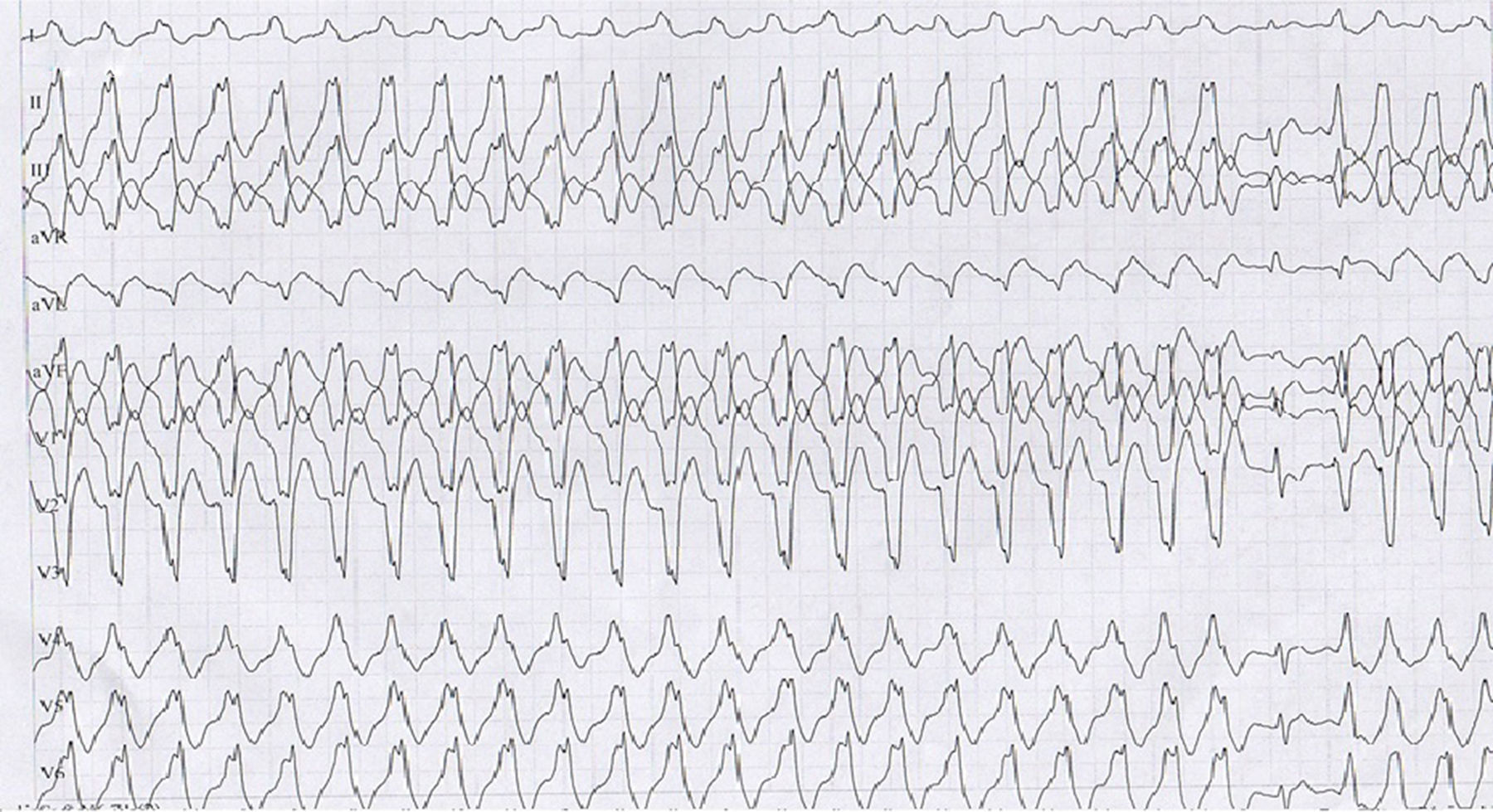
Presentación de casoMujer de 23 años procedente de Socorro, Santander, con clínica iniciada hace 6 años durante el primer mes de embarazo, consistente en episodios de palpitaciones y dolor torácico recurrentes exacerbados con actividad física, asociados con síncope y cefalea. En el último mes hubo aumento de la sintomatología por lo que se hizo prueba de esfuerzo en primer nivel, presentando taquicardia ventricular (TV) monomórfica sostenida con morfología de bloqueo de rama izquierda (fig. 1) asociada a síncope, la cual se manejó inicialmente con la suspensión de la prueba, con lo cual resolvió la arritmia. Por lo anterior la remitieron a nuestra institución, a la cual ingresó hemodinámicamente estable, asintomática, en ritmo sinusal pero con trastorno de la conducción interventricular (hemibloqueo anterosuperior izquierdo) y signos de isquemia subepicárdica de cara anterior. Por ello se inició el abordaje electrofisiológico con la impresión diagnóstica de una TV a estudio.

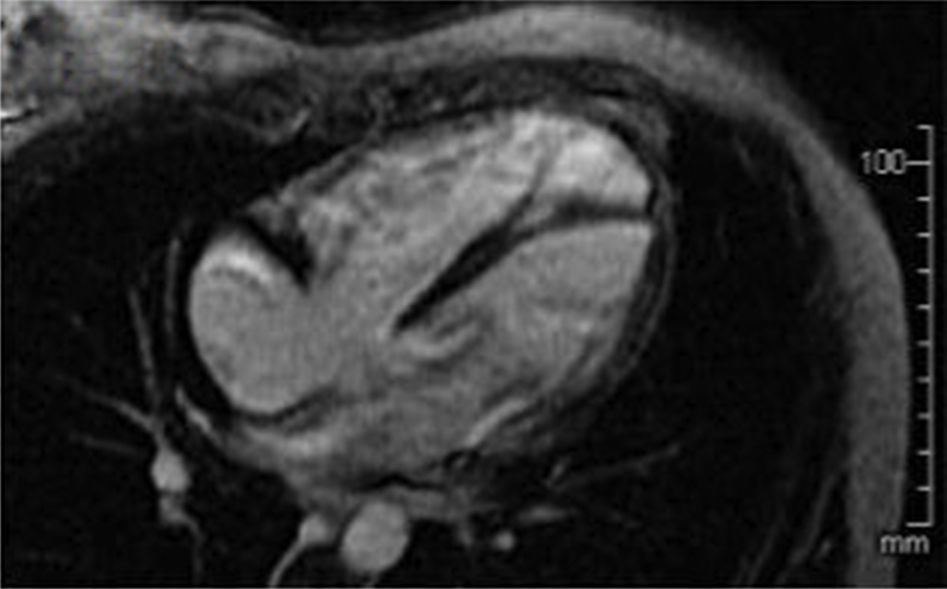
Se le realizó resonancia magnética nuclear cardiaca contrastada (fig. 2), evidenciando dilatación de cavidades cardiacas derechas, especialmente del ventrículo derecho (VD), el cual presentaba aumento en su trabeculación y disminución de su función con una fracción de eyección del 30%. Sin embargo, no se identificaron alteraciones segmentarias en la motilidad de la pared del VD, formaciones aneurismáticas, ni depósitos grasos anómalos o áreas de fibrosis en borde libre del VD.

El ecocardiograma transtorácico mostró VD de tamaño normal, función sistólica y diastólica normal con una fracción de eyección del 60%. Leve crecimiento del eje longitudinal del VD, insuficiencia tricuspídea grado ii con presión sistólica de la arteria pulmonar de 28mmHg.
Inicialmente en junta médica quirúrgica se consideró realizar ablación con mapeo tridimensional y ecografía intracardiaca, realizándose procedimiento con modificación del sustrato arritmogénico de una de las taquicardias a nivel de la pared libre del tracto de salida del VD, pero en la comprobación con infusión de isoproterenol a 2μg/min iv a partir de 100lpm se iniciaron nuevamente episodios de TV no sostenida de diferentes focos con morfología de bloqueo de rama izquierda y un evento de flutter ventricular polimórfico que requirió cardioversión eléctrica sincrónica, por lo que se definió necesidad de ablación percutánea con radiofrecuencia de ganglio estrellado cervical e implante de cardiodesfibrilador (CDI) bicameral como prevención secundaria de MSC, la cual fue realizada sin complicaciones. La estrategia del CDI usada fue una zona de monitor en 150lpm, dos zonas de TV en 167 y 188lpm y una zona de fibrilación ventricular en 200lpm.
De acuerdo con los diferentes estudios diagnósticos y los hallazgos clínicos reportados en este caso, en un principio la primera impresión diagnóstica fue una probable TV polimórfica catecolaminérgica con origen biventricular y se le dio el alta a la paciente 6 días después de la realización del procedimiento, se le indicó continuar tratamiento con metoprolol, omeprazol y acetaminofén, además cambios en el estilo de vida y control con electrofisiología y cardiología.
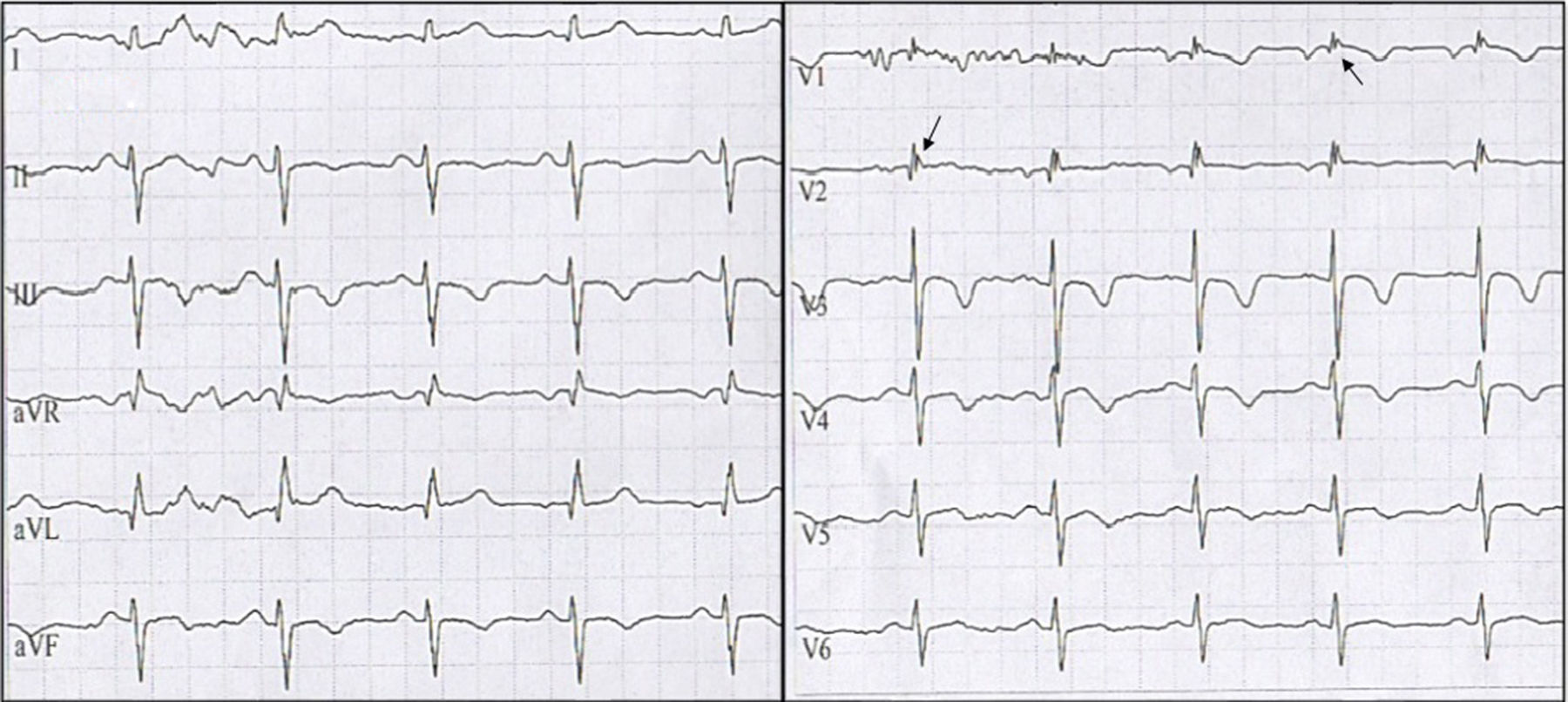
Tres meses después consultó nuevamente al servicio de urgencias por múltiples descargas del dispositivo asociadas a palpitaciones. Al ingreso con electrocardiograma (ECG) con ritmo sinusal de base, extrasístoles ventriculares frecuentes y trastorno de repolarización en cara anteroseptal, por lo cual se realizó telemetría del CDI donde se documentaron ocho episodios de TV con seis descargas apropiadas, además de un evento de fibrilación auricular paroxística sin descarga. Se adicionó al manejo amiodarona y se realizó nuevo ecocardiograma, encontrando VD levemente dilatado principalmente en el tracto de salida del VD, cambio de área fraccional de 0.34 con función discretamente disminuida, diámetro basal de 38mm, medio de 37mm, longitudinal de 74mm, hipertrabeculación apical con desarreglo de la banda moderadora, adelgazamiento de la pared libre en tercio medio y apical, y adicionalmente impresionó discinesia en la punta del ápex. Se revisó nuevamente el ECG que mostró onda anormal al final de QRS lo cual era compatible con ondas épsilon, además de alteraciones de la conducción interventricular y repolarización miocárdica (fig. 3). Dada la evolución de la paciente se realizó una revisión exhaustiva de historia clínica encontrando que la paciente tenía características que podían indicar M/DAVD por lo cual se aplicaron los criterios de Task Force concluyendo que al cumplir con más de 2 criterios mayores, la paciente presentaba un diagnóstico definitivo de esta enfermedad, por lo cual se indicaron estudios genéticos los cuales se encuentran pendientes a la fecha.
Revisión de temaDefinición e historia
La M/DAVD es una miocardiopatía genética que afecta principalmente el VD y que puede generar un compromiso biventricular progresivo. Su primera descripción fue hecha por Giovanni Lancisi en su libro Motu Cordis et Aneurysmatibus en 1736, en el cual describía una familia con recurrencia de la enfermedad durante 4 generaciones, los miembros eran jóvenes y presentaban palpitaciones, dilatación del VD, insuficiencia cardiaca y MSC1,2. Posteriormente, en 1905, Osler describió casos de hombres sanos que morían durante la actividad física leve y encontró que tenían adelgazamiento biventricular. Asimismo informes posteriores describieron presentaciones similares a las reportadas por Osler, incluyendo una descripción realizada por Uhl, en la cual se describía una anomalía congénita con aplasia de las paredes libres del VD, este último caso recibió el nombre de enfermedad de Uhl6,7. Sin embargo la primera descripción clínica completa fue realizada en 1982 por Marcus et al., en donde se usó por primera vez el término «displasia arritmogénica del VD» en una serie de 24 pacientes que presentaban taquiarritmia ventricular con morfología de bloqueo de rama izquierda, alteraciones cinéticas de la pared del VD y reemplazo progresivo de tejido cardiaco normal por tejido adiposo y fibroso8,9.
EpidemiologíaSe estima que la prevalencia es alrededor de 1:2,500-1:5,000 en población en general, siendo mayor en el género masculino (3:1). El rango de edad de presentación es variable (entre 12 y 50 años), con una media estimada de 31 años, siendo poco frecuente después de los 60 años1,2,5,7. La M/DAVD es una de las principales causas de MSC debido a taquiarritmias ventriculares, especialmente en población menor de 35 años y/o atletas2,10.
Fisiopatología y genéticaLa M/DAVD es una enfermedad hereditaria de carácter autosómico dominante con penetrancia incompleta y expresividad variable, aunque en algunos casos puede presentar un patrón autosómico recesivo como es el caso de la enfermedad de Naxos y el síndrome de Carvajal5. La enfermedad consiste en un reemplazo del miocardio del VD por tejido fibroadiposo, es un fenómeno progresivo que inicia desde el epicardio y se extiende hasta llegar al endocardio, lo cual implica un debilitamiento de la pared libre, lo que predispone al desarrollo de procesos aneurismáticos y dilatación ventricular. Igualmente la displasia fibroadiposa interfiere con la conducción ventricular, generando retraso en la conducción del impulso eléctrico y aparición de fenómenos de reentrada2–4,7.
Análisis genéticos y moleculares han generado gran entendimiento sobre su fisiopatología, mostrando mutaciones en varios genes que codifican componentes de los desmosomas cardiacos y que pueden estar asociados a M/DAVD, llevando a que las proteínas de adhesión celular tales como la placoglobina, desmoplaquina, placofinina-2 y desmogleina-2 sean deficientes. Sin embargo, el mecanismo por el cual las mutaciones en los genes de los desmosomas causan la enfermedad no está del todo claro; algunas hipótesis plantean que la pérdida de la función de los desmosomas produce un desacople de los discos intercalados del miocardiocito y una alteración en la adhesión célula-célula. Igualmente se han descrito mutaciones en genes no relacionados con los desmosomas, lo cual podría explicar la gran variabilidad fenotípica de esta enfermedad como es el caso de variaciones en el receptor RYR2 que se asocian al desarrollo de TV polimórfica catecolaminérgica; y alteraciones del factor de crecimiento transformador-B3 involucrado en la producción de citoquinas estimulantes de fibrosis y modulación de las células de adhesión1,5,9,11. Por otro lado, algunos autores indican que estaría implicada una infección viral como detonante de la patogénesis de la enfermedad en pacientes genéticamente predispuestos, además de procesos inflamatorios los cuales son hallazgos comunes en autopsias y biopsias de estos pacientes2,5,12–15.
Manifestaciones clínicasLa sintomatología suele presentarse en pacientes entre la segunda y quinta decada de la vida, en su mayoría previamente sanos1. Los síntomas pueden estar correlacionados con arritmias ventriculares y suelen aparecer con el esfuerzo. Los síntomas más frecuentes son el síncope (27%), palpitaciones (26%), MSC (23%), mareo e insuficiencia cardiaca que se presenta en menos del 10% de los pacientes1,5,6,11,16. Además la insuficiencia cardiaca sumada a una fibrilación auricular (presente en un 25% de los pacientes), puede generar trombosis mural endocavitada en los aneurismas ventriculares o en los apéndices auriculares causando procesos de tromboembolismo2,17.
La historia natural de la enfermedad clásicamente se ha dividido en: 1) Una fase subclínica con anormalidades estructurares ocultas, en la que no hay manifestaciones clínicas aparentes pero el paciente está en riesgo de MSC. 2) Una alteración eléctrica sintomática, en la que el paciente presenta síncope y palpitaciones por arritmias generalmente provocadas con esfuerzo y anormalidades morfológicas del VD evidentes en imágenes convencionales. 3) Insuficiencia del VD por la pérdida progresiva del miocardio. 4) Por último, insuficiencia biventricular, que es semejante a una miocardiopatía dilatada. Sin embargo, los pacientes no necesariamente presentan esa secuencia2–5.
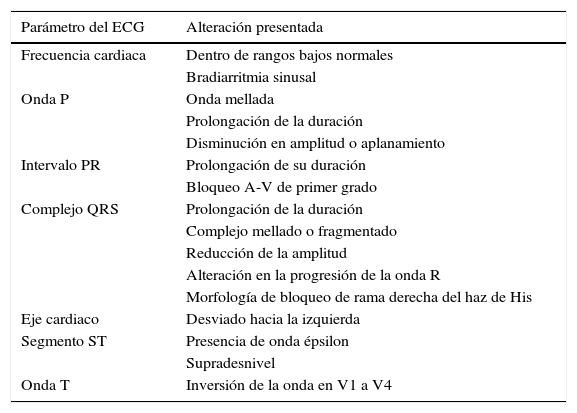
El ECG es una herramienta importante en la evaluación de estos pacientes, se deben observar las 12 derivaciones y tener en cuenta la edad, el sexo, la medicación actual y/o la terapia con dispositivos cardiacos. En la tabla 1 se muestran las principales alteraciones electrocardiográficas asociadas a M/DAVD. A pesar de que las manifestaciones electrocardiográficas son de utilidad clínica y diagnóstica, ninguna se considera completamente específica12,18. En un estudio realizado por Saguner et al. se concluyó que la M/DAVD es una enfermedad progresiva y que esta progresión puede ser evidente en el ECG, por lo que se ha planteado la posibilidad de su uso como evaluador de la progresión de la enfermedad13.
Alteraciones más frecuentes en el ECG de pacientes con M/DAVD
| Parámetro del ECG | Alteración presentada |
|---|---|
| Frecuencia cardiaca | Dentro de rangos bajos normales |
| Bradiarritmia sinusal | |
| Onda P | Onda mellada |
| Prolongación de la duración | |
| Disminución en amplitud o aplanamiento | |
| Intervalo PR | Prolongación de su duración |
| Bloqueo A-V de primer grado | |
| Complejo QRS | Prolongación de la duración |
| Complejo mellado o fragmentado | |
| Reducción de la amplitud | |
| Alteración en la progresión de la onda R | |
| Morfología de bloqueo de rama derecha del haz de His | |
| Eje cardiaco | Desviado hacia la izquierda |
| Segmento ST | Presencia de onda épsilon |
| Supradesnivel | |
| Onda T | Inversión de la onda en V1 a V4 |
ECG: electrocardiograma; M/DAVD: miocardiopatía/displasia arritmogénica ventricular derecha.
Fuente: Zhang et al.12.
Debido a su amplio espectro de presentación clínica, en la actualidad la M/DAVD se diagnostica mediante los criterios modificados en el 2010 por la Task Force, que incluyen 6 categorías, cada una con criterios mayores y menores19 (tabla 2.). El diagnóstico definitivo se hace con 2 criterios mayores o un criterio mayor y 2 menores, o 4 criterios menores de categorías diagnósticas diferentes. Un diagnóstico limítrofe se hace con un criterio mayor y un criterio menor, o 3 criterios menores de categorías diagnósticas diferentes. Un diagnóstico posible se hace con un criterio mayor o 2 criterios menores de categorías diagnósticas diferentes1,2,5–7,19.
Criterios de 2010 de la Task Force para el diagnóstico de la miocardiopatía/displasia arritmogénica del ventrículo derecho
| I. Disfunción y alteraciones estructurales globales o regionales | |
|---|---|
| Mayores | En el ecocardiograma bidimensional: acinesia, discinesia o aneurisma regionales del VD y una de las siguientes (en telediástole): • PELP TSVD≥32mm (corregido por tamaño corporal [PELP/ASC]≥19mm/m2) • PECP TSVD≥36mm (corregido por tamaño corporal [PECP/ASC]≥21mm/m2) • O cambio del área fraccional≤33% |
| En la RM: acinesia o discinesia regionales del VD o contracción disincrónica del VD y una de las siguientes: • Cociente de volumen telediastólico del VD respecto a ASC≥110ml/m2 (varones) o≥100ml/m2 (mujeres) • O fracción de eyección del VD≤40% | |
| En la angiografía del VD: acinesia, discinesia o aneurisma regionales del VD | |
| Menores | En el ecocardiograma bidimensional: acinesia o discinesia regionales del VD y una de las siguientes (en el periodo telediastólico): • PELP TSVD≥29 a<32mm (corregido por tamaño corporal [PELP/ASC]≥16 a<19mm/m2) • PECP TSVD≥32 a<36mm (corregido por tamaño corporal [PECP/ASC]≥18 a<21mm/m2) • O cambio del área fraccional>33 a≤40% |
| En la RM: acinesia o discinesia regionales del VD o contracción disincrónica del VD y una de las siguientes: • Cociente de volumen telediastólico del VD respecto a ASC≥100 a<110ml/m2 (varones) o≥90 a<100ml/m2 (mujeres) • O fracción de eyección del VD>40 a≤45% |
| II. Caracterización del tejido de la pared | |
|---|---|
| Mayores | Miocitos residuales<60% mediante análisis morfométrico (o<50% si se han estimado), con sustitución fibrosa del miocardio de la pared libre del VD en al menos una muestra, con o sin sustitución adiposa del tejido en la biopsia endomiocárdica |
| Menores | Miocitos residuales del 60-75% mediante análisis morfométrico (o del 50-65% si se han estimado), con sustitución fibrosa del miocardio de la pared libre del VD en al menos una muestra, con o sin sustitución adiposa del tejido en la biopsia endomiocárdica |
| III. Anomalías de la repolarización | |
|---|---|
| Mayores | Ondas T invertidas en las derivaciones precordiales derechas (V1, V2 y V3) o más allá en individuos de edad>14 años (en ausencia de BRDH con QRS≥120ms) |
| Menores | • Ondas T invertidas en las derivaciones V1 y V2 en individuos de edad>14 años (en ausencia de BRDH completo) o en V4, V5 o V6. • Ondas T invertidas en las derivaciones V1, V2, V3 y V4 en individuos de edad>14 años en presencia de un BRDH completo |
| IV. Anomalías de despolarización/conducción | |
|---|---|
| Mayores | Onda épsilon (señales de baja amplitud reproducibles entre el final del complejo QRS y el inicio de la onda T) en las derivaciones precordiales derechas (V1 a V3) |
| Menores | • Potenciales tardíos mediante SAECG en al menos uno de 3 parámetros, en ausencia de una duración del QRS≥110ms en el ECG estándar: duración del QRS filtrado≥114ms; duración del QRS terminal<40μV (duración de señal de baja amplitud)≥38ms; raíz de la media cuadrados de los voltajes de los 40ms terminales 20μV • Duración de la activación terminal del QRS≥55ms medida desde el mínimo de la onda S hasta el final del QRS, incluyendo R’, en V1, V2 o V3, en ausencia de BRDH completo |
| V. Arritmias | |
|---|---|
| Mayores | Taquicardia ventricular no sostenida o sostenida con morfología de BRIH con eje superior (QRS negativo o indeterminado en las derivaciones ii, iii, aVF; y positivo en aVL) |
| Menores | Taquicardia ventricular no sostenida o sostenida de configuración de TSVD, con morfología de BRIH con eje inferior (QRS positivo en las derivaciones ii, iii y aVF y negativo en aVL) o de eje desconocido>500 extrasístoles ventriculares por 24h (Holter) |
| VI. Antecedentes familiares | |
|---|---|
| Mayores | • M/DAVD confirmada en un familiar de primer grado que cumpla los criterios actuales de la Task Force • M/DAVD confirmada anatomopatológicamente en la autopsia o la intervención quirúrgica en un familiar de primer grado • Identificación de una mutación patogénica clasificada como asociada o probablemente asociada a la M/DAVD en el paciente examinado |
| Menores | • Antecedentes de M/DAVD en un familiar de primer grado en el que no es factible determinar si cumple los criterios actuales de la Task Force • Muerte súbita prematura (<35 años de edad) debida a presunta M/DAVD en un familiar de primer grado • M/DAVD confirmada anatomopatológicamente o mediante los criterios actuales de la Task Force en un familiar de segundo grado |
ASC: área de superficie corporal; aVF: aumento de voltaje en derivación unipolar del pie izquierdo; aVL: aumento de voltaje en derivación unipolar de brazo izquierdo; BRDH: bloqueo de rama derecha del haz; BRIH: bloqueo de rama izquierda del haz; ECG: electrocardiograma; M/DAVD: miocardiopatía/displasia arritmogénica ventricular derecha; PECP: proyección de eje corto paraesternal; PELP: proyección de eje largo para esternal; RM: resonancia magnética; SAECG: electrocardiograma de promediación de señal; TSVD: tracto de salida del ventrículo derecho; VD: ventrículo derecho.
Modificada de Quarta y Elliot19.
Los objetivos más importantes del manejo clínico de estos pacientes incluyen: 1) reducir la mortalidad a causa de MSC o falla cardiaca; 2)prevención de la progresión de la enfermedad; 3) disminuir los síntomas y mejorar la calidad de vida de los pacientes y 4) limitar los síntomas de falla cardiaca y mejorar la capacidad funcional de los pacientes20. Las estrategias terapéuticas consideran la modificación en el estilo de vida, que incluye restricción de ejercicio, tratamiento con fármacos antiarrítmicos, colocación de CDI, ablación con catéter, ablación por radiofrecuencia y trasplante cardiaco5,21.
Restricción de ejercicioSe ha demostrado que el ejercicio de resistencia puede predisponer a una presentación más temprana de la enfermedad, cursando esta con manifestaciones clínicas más severas dadas por arritmias de aparición más temprana, disfunción estructural, falla cardiaca y necesidad de trasplante1,2,4,9,15.
La recomendación actual por la AHA y el Colegio Americano de Cardiología para pacientes con M/DAVD probable, limítrofe o definitiva es limitar la actividad física a actividades con baja demanda cardiovascular, como caminar. No se ha documentado una frecuencia cardiaca o nivel de esfuerzo «seguro» y se necesitan más estudios para aclarar las restricciones adecuadas para los pacientes y sus familias1,9,20.
Fármacos antiarrítmicosEl objetivo del tratamiento con fármacos antiarrítmicos es mejorar la calidad de vida mediante la prevención de arritmias ventriculares sintomáticas y descargas del CDI1,20. En pacientes que presentan episodios de arritmias sin descompensación hemodinámica, los betabloqueadores y antiarrítmicos de clase iii, solos o en combinación, han demostrado ser los agentes más efectivos en la supresión de las arritmias ventriculares con un bajo efecto proarritmogénico. Sin embargo, es difícil evaluar la terapia antiarrítmica ya que los pacientes tienden a tener múltiples eventos arrítmicos a lo largo del tiempo y los medicamentos son cambiados con frecuencia, por lo que no hay ensayos aleatorios controlados para comparar los diferentes fármacos antiarrítmicos disponibles. Pero la amiodarona y el sotalol son los que se usan con más frecuencia1,8,14,20,22.
Algunos estudios retrospectivos indican que la terapia farmacológica no debe considerarse como terapia primaria para prevenir la MSC y que su uso se debe limitar a la terapia adyuvante en pacientes con arritmias ventriculares sostenidas tratados con un CDI21. Además no se recomienda el tratamiento en pacientes asintomáticos sin arritmias ventriculares documentadas20.
Cardiodesfibrilador implantableLos datos disponibles de estudios observacionales concluyen que el CDI es una terapia eficaz en la prevención de MSC y segura en pacientes con M/DAVD23–26. Sin embargo, después de establecido el diagnóstico de M/DAVD, se debe tomar la decisión de si se va a colocar un CDI4,9. Por un lado, la esperanza de vida de esta población joven es cercana a lo normal si la MSC se previene. Por otra parte, la implantación del CDI a una edad joven compromete al paciente a múltiples sustituciones de los generadores y un riesgo acumulado significativo de falla del dispositivo, choques inapropiados e infección21.
El implante del CDI se recomienda en pacientes con M/DAVD que hayan experimentado uno o más episodios de TV sostenida o fibrilación ventricular hemodinámicamente inestables. Además en aquellos pacientes con disfunción sistólica severa del VD o biventricular independiente de la arritmia que presente, o que tengan factores de riesgo como síncope de origen desconocido, disfunción ventricular moderada o TV no sostenida, potenciales tardíos, onda épsilon y antecedente familiar1,15,20. Actualmente no se recomienda el implante profiláctico de CDI en pacientes asintomáticos debido al pronóstico general favorable y al elevado riesgo de complicaciones relacionadas con el dispositivo1,2,4,20,21.
Ablación con catéterLas indicaciones para la ablación con catéter en pacientes con M/DAVD incluyen TV monomórfica bien tolerada con formas localizadas de la enfermedad, TV sostenida refractaria a antiarrítmicos y descargas frecuentes del CDI. Las técnicas de mapeo y ablación actuales incluyen el mapeo de arrastre durante la TV tolerada y la ablación usando sistemas de mapeo electroanatómicos tridimensionales1,5,9,11. Es importante reconocer que este procedimiento no es curativo sino que mejora la calidad de vida mediante la disminución de la frecuencia de episodios de TV1,9,20.
Ablación por radiofrecuenciaLa ablación por radiofrecuencia de un foco activo es una terapia alternativa que se recomienda en pacientes con TV que no responden a terapia farmacológica y en pacientes con CDI que sufren de arritmias ventriculares recurrentes, con descargas del dispositivo2,4,7. Por otro lado, no está contemplado su uso como alternativa del CDI para la prevención de MSC20.
El procedimiento tiene una tasa de éxito agudo entre el 60 y 90%, pero las recidivas son frecuentes debido a la naturaleza progresiva de la enfermedad, con una tasa de TV recurrente hasta del 90% a 3 años, usualmente por un nuevo foco que toma el control5,7,9. Se ha propuesto que un enfoque combinado de ablación y mapeo endocárdico y epicárdico mejoran los resultados3–5,20.
Trasplante cardiacoEl trasplante cardiaco se recomienda como última opción terapéutica en pacientes con insuficiencia cardiaca congestiva severa que no responden a tratamiento óptimo o episodios recurrentes de TV o fibrilación ventricular refractarios a la ablación por catéter o terapia con CDI1,8,9,20,27.
DiscusiónLa relevancia de este caso clínico está dada por la baja prevalencia y pocos reportes de casos en la literatura latinoamericana por lo cual no existen datos exactos de su epidemiología2. Pese a lo anterior se reconoce que la prevalencia de la M/DAVD oscila entre 1:2,500 y 1:5,000 en población general. Sin embargo, existe un subdiagnóstico de esta entidad debido a la gran variedad de presentaciones clínicas no reconocidas y a que la mayoría de estos pacientes por lo general se diagnostican cuando presentan cuadros severos y posterior a la exclusión de otras entidades como fue el caso de nuestra paciente1,2,4,6.
Se sabe que esta enfermedad tiene un alto componente hereditario, presente hasta en un 50% de los casos5. Sin embargo, en nuestro caso, no se lograron establecer condiciones similares en los familiares interrogados. Igualmente es de resaltar que el inicio de la sintomatología se dio durante el primer trimestre del embarazo, lo cual lleva a plantear la posibilidad de una asociación entre el embarazo y el desarrollo de la M/DAVD, ya que durante el embarazo el volumen plasmático, la frecuencia y el gasto cardiaco aumentan, lo cual conduce a cambios hemodinámicos no predecibles que pueden condicionar el desarrollo de los síntomas; si bien no es común esta asociación, se han descrito casos en la literatura que nos permiten plantear esta posibilidad28–31.
La presentación clínica inicial de nuestra paciente, a pesar de que es típica para la enfermedad no es específica para la misma, por ello en el proceso diagnóstico se consideraron otras enfermedades como la TV catecolaminérgica, cuyo mecanismo más aceptado es el de actividad gatillada con posdespolarizaciones tardías, motivo por el cual se realizó la prueba con isoproterenol como inductor de TV, pese a que la M/DAVD tiene un mecanismo de reentrada. Sin embargo, ante la no mejoría clínica (probablemente debido a los múltiples focos arritmogénicos presentes, característicos de la M/DAVD) y posterior a la revisión de las pruebas de función mecánica cardiaca se evidenció una insuficiencia estructural y funcional del VD sin compromiso del ventrículo izquierdo. Además por hallazgos de la onda épsilon y alteraciones en la repolarización y conducción miocárdica dentro del ECG, se planteó la posibilidad de una M/DAVD.
Finalmente se llega al diagnóstico definitivo con 4 criterios mayores de la Task Force para M/DAVD dados por: 1) Disfunción regional del VD con FEVD<40%, 2) Onda épsilon en derivación V1-V2 en el ECG, 3) Presencia de TV sostenida con imagen de bloque de rama izquierda durante el test de ergometría y 4) Inversión de ondas T en las derivaciones V1-V4 del ECG19. Dentro del tratamiento se indicaron las medidas recomendadas en la literatura, las cuales incluyen medidas no farmacológicas en el estilo de vida como la no realización de ejercicio moderado a intenso; fármacos antiarrítmico e implante de CDI con seguimiento periódico por el servicio de electrofisiología20. Esto nos lleva a concluir la importancia de los estudios diagnósticos e intervención temprana en el paciente joven con síncope y arritmia ventricular.
FinanciaciónNo se recibió financiamiento para el desarrollo de este artículo.
Conflicto de interesesLos autores declaran no tener conflictos de intereses.