




Un varón de 4 años, sin antecedentes relevantes, consulta por disminución de agudeza visual bilateral, más acusada en condiciones escotópicas, que no mejora con corrección óptica. No se aprecian alteraciones fundoscópicas significativas, por lo que se sospecha una distrofia retiniana. La secuenciación del gen CACNA1F detecta la mutación c.3081C>A (p.Tyr1027Ter), que se ha producido de novo en la madre del paciente. Esta mutación, en el contexto clínico referido y con un patrón electronegativo compatible, establece el diagnóstico de ceguera nocturna estacionaria congénita tipo 2 ligada al cromosoma X.
La electrofisiología y el estudio genético deben formar parte del protocolo diagnóstico de cualquier pérdida de visión inexplicada en niños. La descripción, la nomenclatura y la clasificación de las distrofias retinianas hereditarias con base en sus características genotípicas y electrorretinográficas evita los errores diagnósticos derivados de su habitual superposición clínica y fenotípica.
A 4-year-old boy, with no history of relevance, presented with bilateral visual impairment, more so in scotopic conditions, and did not improve with optical correction. No significant funduscopic abnormalities were seen, leading to a suspicion of retinal dystrophy. Sequencing of the CACNA1F gene detected the c.3081C>A (p.Tyr1027Ter) mutation, which had occurred de novo in the patient's mother. This mutation, in the aforementioned clinical context, and with a compatible electronegative pattern, establishes the diagnosis of X-linked type 2 congenital stationary night blindness.
Electrophysiology and genetic testing should be part of the diagnostic protocol for any unexplained loss of vision in children. The description, nomenclature and classification of hereditary retinal dystrophies based on their genotypic and electroretinograpic characteristics, avoids diagnostic errors due to their usual clinical and phenotypic overlap.
La ceguera nocturna estacionaria congénita (CSNB) hace referencia a un grupo muy poco frecuente de trastornos hereditarios de inicio en la infancia, que se caracterizan por alteraciones en la visión escotópica y disfunción retiniana no progresiva. Los primeros pacientes descritos con CSNB fueron los descendientes de Jean Nougaret, que nació en 1637 en el sur de Francia. El espectro de la CSNB es amplio e incluye variantes con distintos patrones de herencia y diferente expresión fenotípica; no obstante, el aspecto del fondo de ojo es normal en la mayoría de sus formas. Presentamos este caso clínico con la intención de recordar que el electrorretinograma (ERG) y la secuenciación génica son fundamentales en el estudio de enfermedades como la CSNB, de gran heterogeneidad clínica y muy baja incidencia que, de lo contrario, pueden sufrir de una importante demora diagnóstica o que incluso llegan a ser etiquetadas como otras enfermedades1.
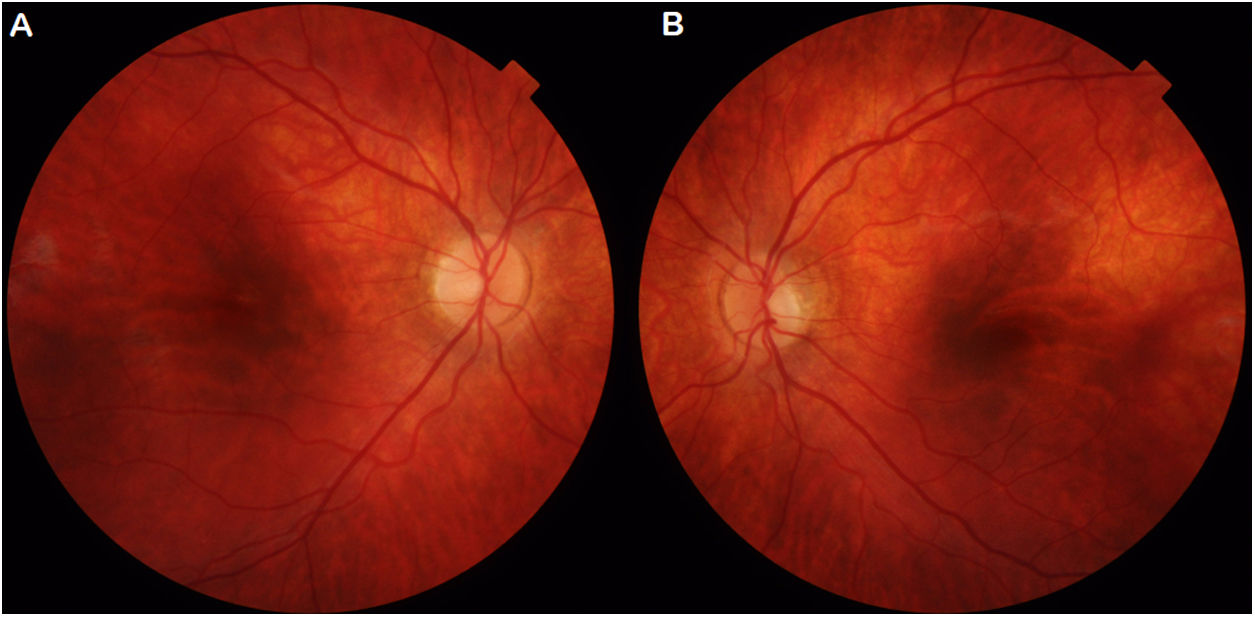
Caso clínicoUn varón caucásico de 4 años, sin antecedentes personales ni familiares de interés, consulta por disminución de agudeza visual (AV) bilateral, más acusada en condiciones escotópicas. Sus padres son sanos y no consanguíneos. El paciente presenta una ambliopía profunda, con AV de 20/100 en el ojo derecho (OD) y de 20/200 en el ojo izquierdo (OI), con supresión de OI en binocularidad y nula visión estereoscópica. La refracción ciclopléjica evidencia un astigmatismo hipermetrópico bilateral poco significativo, 120° + 1,00 + 2,00 en el OD y 60° + 1,25 en el OI. No se aprecian alteraciones pupilares, estrabismo, nistagmo ni defectos de transiluminación del iris. El paciente realiza el test de Ishihara y, posteriormente, el test de Farnsworth, sin confirmarse la existencia de anomalías cromáticas en ninguno de ambos, a pesar de su excelente colaboración. Sin embargo, los campos visuales solicitados no son valorables. El fondo de ojo es ligeramente hipopigmentado a nivel peripapilar y en arcadas temporales, en ausencia de otros signos de patología fundoscópica (fig. 1). Además, la tomografía de coherencia óptica no revela signos de afectación papilar o foveal. Se prescribe corrección óptica, pero el paciente apenas experimenta mejoría en su AV, por lo que, ante la sospecha de una distrofia retiniana, se solicitan pruebas electrofisiológicas y estudio genético.
. Disco óptico levemente oblicuo en el OI (B).")
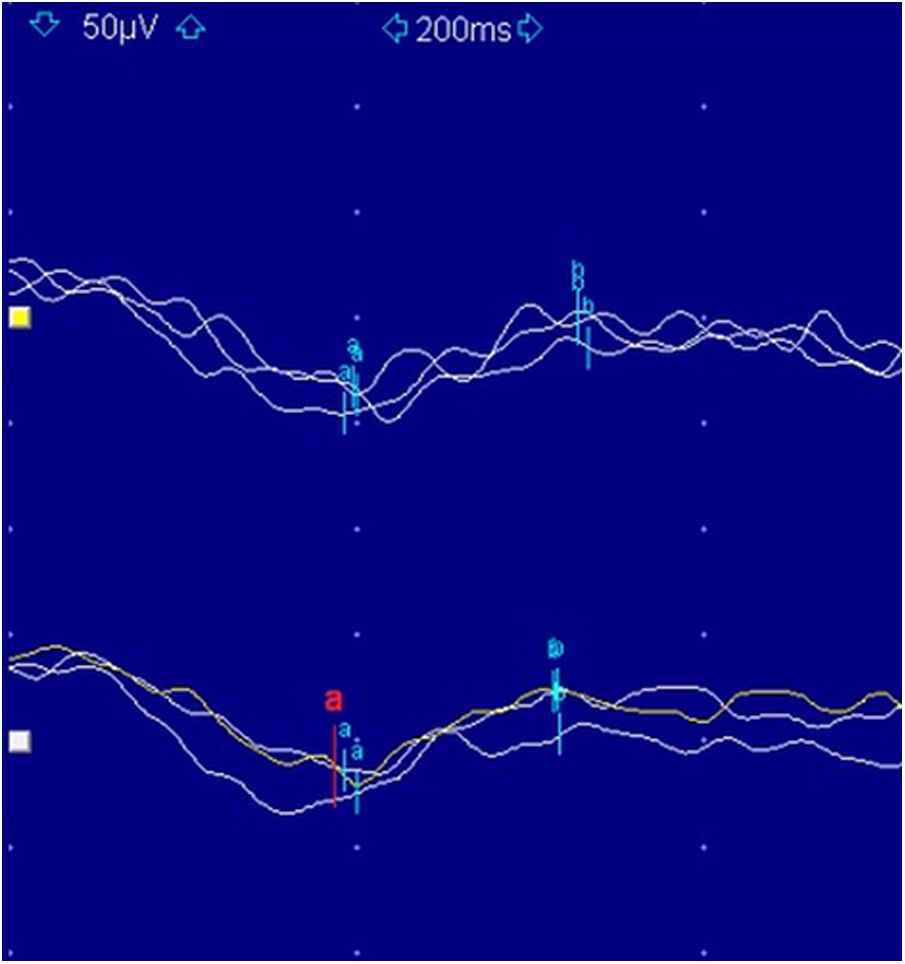
El estudio electrofisiológico consta de unos potenciales evocados visuales tipo pattern, con resultados normales, y de un ERG flash, con aumento de latencias y disminución de amplitudes en ambos ojos. Estos hallazgos del ERG sugieren una afectación de la retina periférica; no obstante, su valor diagnóstico en edades tan tempranas es limitado debido a su incapacidad para discriminar entre respuestas fotópicas y escotópicas. Por esta razón, a los 6 años, se realiza un ERG de campo completo que muestra un patrón electronegativo (fig. 2), alteración de las respuestas fotópicas y escotópicas, así como de los potenciales oscilatorios.
. Patrón electronegativo, con reducción de la amplitud de la onda b en relación con la onda a, en la respuesta combinada de conos y bastones en condiciones escotópicas.")
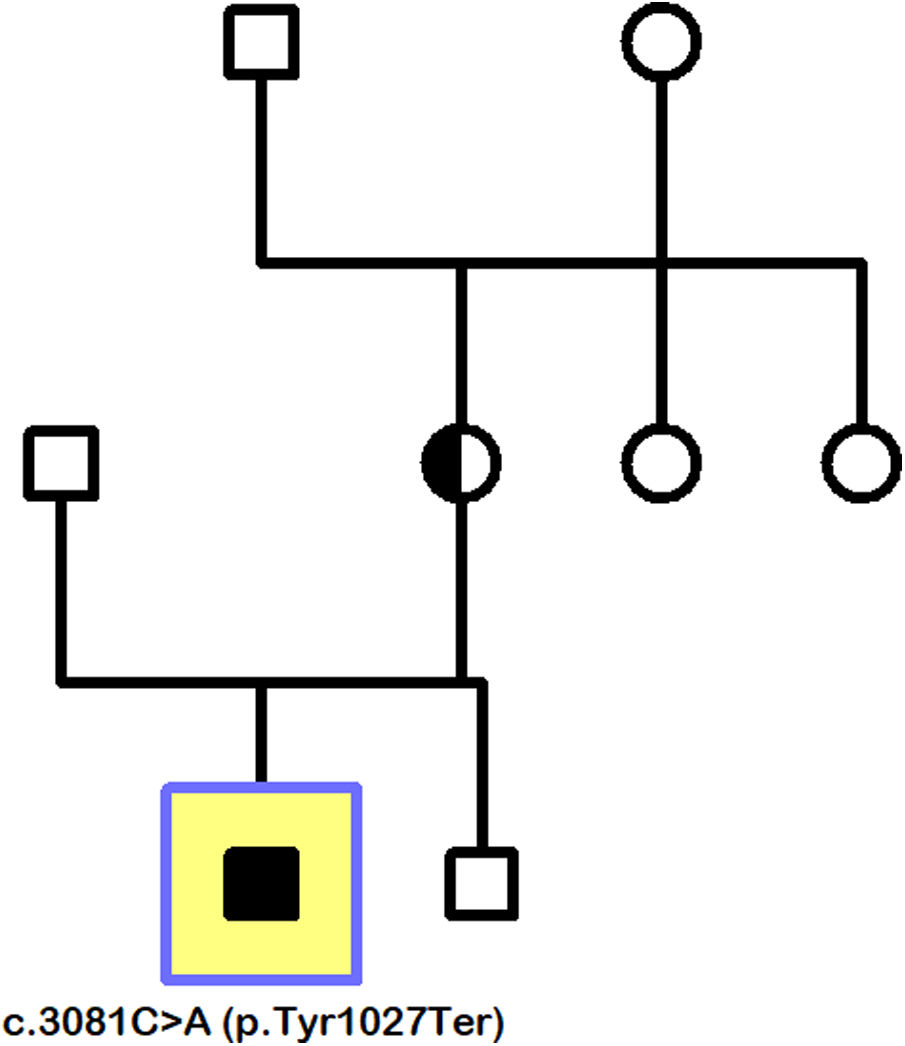
La secuenciación génica se orienta a las distrofias de conos y bastones, y evidencia la variante patogénica c.3081C>A (p.Tyr1027Ter) en el gen CACNA1F situado en Xp11.23. La presencia de esta mutación, así como un cuadro clínico y un ERG compatibles, establecen el diagnóstico de CSNB tipo 2 (CSNB2). El estudio familiar demuestra que la madre del paciente es portadora de una mutación truncante de novo, que no proviene de los abuelos maternos y que se ha transmitido a su hijo mediante herencia recesiva ligada al cromosoma X. Por su parte, el hermano menor del paciente no ha heredado la mutación y, por tanto, no ha resultado afectado por la enfermedad (fig. 3).
. Madre portadora de la mutación de novo c.3081C>A (p.Tyr1027Ter), transmitida a su hijo mediante herencia recesiva ligada al cromosoma X. Ausencia de la mutación en el hermano varón del paciente índice, así como en los abuelos maternos, lo que hace innecesario ampliar el estudio a otros miembros de su rama familiar.")
Pedigrí familiar (paciente índice señalado por cuadrado). Madre portadora de la mutación de novo c.3081C>A (p.Tyr1027Ter), transmitida a su hijo mediante herencia recesiva ligada al cromosoma X. Ausencia de la mutación en el hermano varón del paciente índice, así como en los abuelos maternos, lo que hace innecesario ampliar el estudio a otros miembros de su rama familiar.
Actualmente, a los 8 años de edad, la mejor AV corregida es de 20/30 en el OD (120° + 1,50 + 1,25) y de 20/40 en el OI (85° + 1,00), con buena fusión binocular y una estereopsis parcial de 240”.
DiscusiónEl patrón hereditario más frecuente en la CSNB es el recesivo ligado al cromosoma X, habiéndose descrito al menos 17 genes implicados, con más de 360 mutaciones posibles y más de 670 alelos afectados. Estos genes codifican proteínas involucradas en la fototransducción. Los casos esporádicos de CSNB suponen aproximadamente el 40%1.
La heterogeneidad clínica de la CSNB es significativa, con grados variables de déficit visual y de nictalopía. La mayoría de las variantes de la enfermedad (a excepción de la enfermedad de Oguchi y el fundus albipunctatus) no presentan alteraciones fundoscópicas. La AV depende en gran medida de los frecuentes errores refractivos asociados, en especial de la miopía. Los pacientes con CSNB recesiva ligada al cromosoma X se asocian más habitualmente con miopía elevada y con baja AV. Algunos casos se acompañan de nistagmo o estrabismo. La nictalopía no siempre es sintomática, seguramente como consecuencia de una respuesta funcional adaptativa, lo que motiva que, en muchas ocasiones, la afectación de la visión escotópica pase desapercibida y esta enfermedad sea infradiagnosticada1. La microperimetría demuestra una reducción significativa de la sensibilidad macular en estos pacientes, en comparación con controles sanos2. Las mujeres portadoras son casi siempre asintomáticas, aunque pueden mostrar cambios evidentes en el ERG (ausencia de potenciales oscilatorios)3.
El ERG de campo completo clasifica funcionalmente la CSNB en 2grupos. El tipo Riggs es muy poco habitual y se corresponde casi exclusivamente con las formas autosómicas dominantes, en las cuales se observa una alteración en las respuestas escotópicas (onda b exagerada en ausencia de onda a), con normalidad de las respuestas fotópicas, lo que sugiere un defecto intrínseco de los bastones1. El tipo Schubert-Bornschein aparece asociado a las formas de herencia autosómica recesiva o ligada al cromosoma X y es mucho más frecuente. Se caracteriza este segundo grupo por un patrón electronegativo (pérdida selectiva de la onda b con conservación de la onda a), con alteración mixta de las respuestas fotópicas y escotópicas, lo cual se traduce en una anomalía postransduccional, sin estar afectada directamente la función de conos ni bastones4.
El equipo de Miyake ha estudiado los mecanismos de fototransducción en la CSNB con ERG tipo Schubert-Bornschein y ha descrito 2 vías de disfunción sináptica entre los fotorreceptores y las células bipolares, afirmando que no deben ser consideradas como variantes de una misma enfermedad, si no como entidades clínicas independientes, con mutaciones genéticas, mecanismos etiopatogénicos y patrones electrorretinográficos diferentes5. Los términos empleados en la nomenclatura internacional son los de tipo 1 (CSNB1) o formas completas (cCSNB) y tipo 2 (CSNB2) o formas incompletas (icCSNB)1.
La mutación más común en la CSNB1 o cCSNB afecta al gen NYX, que codifica la nictalopina. Se caracteriza por una disfunción postsináptica que se corresponde con un ERG negativo en condiciones escotópicas, con una función fotópica normal o levemente subnormal. La CSNB1 se asocia con frecuencia con miopía elevada, nistagmo y nictalopía sintomática1,5.
La CSNB2 o la icCSNB se asocia con diferentes mutaciones genéticas, siendo la más frecuente la del gen CACNA1F, presente en aproximadamente el 55% de los casos de CSNB ligada al cromosoma X y que se localiza en el brazo corto del cromosoma X (Xp11.23), codificando una proteína transmembrana de un canal de calcio (Cav1.4) que regula la liberación de glutamato en la fototransducción6. La expresión fenotípica de la CSNB2 es más heterogénea, con nictalopía inconstante (54% no refieren alteraciones en su calidad de vida), errores refractivos variables desde miopía a hipermetropía, fotofobia, sensibilidad a la luz, y, en ocasiones, disminución de AV en condiciones fotópicas7. La CSNB2 determina un defecto a nivel presináptico, observándose un ERG con onda b de amplitud disminuida tanto en condiciones escotópicas como fotópicas8. La respuesta fotópica está mucho más afectada que en la CSNB1 y, por ello, la CSNB2 debe ser diferenciada mediante ERG de las distrofias puras de conos, en las cuales suele existir una respuesta escotópica normal1.
La mutación del gen CACNA1F se encuentra asociada no solo a CSNB2, sino también al desarrollo de otras 2patologías oculares: distrofia de conos y bastones ligada al cromosoma X tipo 3 y enfermedad ocular de las islas Åland o síndrome de Forssius-Eriksson. La influencia de modificadores genéticos o ambientales puede ser responsable de su diferente expresión fenotípica9. La normalidad de la visión cromática, la ausencia de nistagmo e hipoplasia foveal y el patrón compatible de ERG apoyan en este caso el diagnóstico de CSNB2. La significación patogénica de la mutación c.3081C>A (p.Tyr1027Ter) en CSNB2 parece no haber sido descrita previamente en la literatura, o al menos, no ha sido encontrada en nuestra búsqueda bibliográfica.
La electrofisiología y el estudio genético deben formar parte del protocolo diagnóstico de cualquier pérdida de visión inexplicada en niños. Este caso clínico ilustra a la perfección el reto que pueden suponer enfermedades como la CSNB2. La presencia de una mutación de novo ha complicado aún más el rastreo de posibles casos familiares en la anamnesis. El ERG y la secuenciación génica han sido imprescindibles para confirmar la afectación de la visión escotópica y para descartar otras distrofias retinianas hereditarias con características fenotípicas similares, pero de carácter progresivo y de peor pronóstico, como la retinosquisis juvenil ligada al cromosoma X o la amaurosis congénita de Leber. Por ello, se recomienda que la descripción, la nomenclatura y la clasificación de este tipo de enfermedades se haga siempre con base en sus características genotípicas y electrorretinográficas, evitando así los errores diagnósticos derivados de su habitual superposición clínica y fenotípica1,5,10.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
