Desde los inicios de la cirugía cardiaca con circulación extracorpórea, el daño de isquemia-reperfusión miocárdico ha sido reconocido como uno de los principales causantes de morbimortalidad.
MetodologíaRevisión bibliográfica sobre isquemia-reperfusión miocárdica a nivel experimental y clínico utilizando Pubmed y otras bases y fuentes de datos incluyendo Teseo.
ResultadosSe realiza una revisión actualizada de los aspectos celulares y bioquímicos de la isquemia-reperfusión miocárdica. Se exponen los cambios celulares durante la isquemia, los cambios celulares durante la reperfusión, los radicales libres de oxígeno, la activación del sistema del complemento, la activación de las citocinas proinflamatorias y la interacción neutrófilo-endotelio. Se adjunta una tabla con un listado de fármacos y compuestos que a nivel experimental y clínico que han sido usados para tratar el daño de isquemia-reperfusión miocárdico.
ConclusionesEn el momento actual se sigue investigando de forma activa en este campo y hay estudios clínicos en marcha encaminados a probar la eficacia de fármacos para reducir el daño de isquemia-reperfusión miocárdico. El cirujano cardiaco debe de estar bien informado en nuevos métodos de cardioprotección en cirugía cardiaca.
Since the beginning of the cardiac surgery with cardiopulmonary bypass, myocardial-reperfusion injury has been recognized as one of the leading causes of morbidity and mortality related with this type of surgery.
MethodologyA bibliographic review in Pubmed and other data base including Teseo has been carried out.
ResultsFundaments of the cellular changes during the ischemic time, cellular changes in the reperfusion time, release of oxygen free radicals, complement activation, proinflammatory cytokines activation and neutrophil-endothelium interaction are exposed. A table with a selected list of drugs and compounds used against the myocardial ischemic -reperfusion injury syndrome is included.
ConclusionsAt present, the myocardial reperfusion injury syndrome is an active line of research. There are many trials and studies on investigating new agents .The cardiac surgeon should be well informed in new strategies in myocardial protection in cardiac surgery.
155 words
En los años 50 del siglo pasado, el Dr. John Gibbon revolucionó la historia de la cirugía al realizar y publicar en humanos las primeras operaciones de cirugía cardiaca usando una bomba (Gibbon-IBM Heart-Lung Machine) o máquina de circulación extracorpórea (CEC)1. Poco después, en esa misma década y de forma independiente, los doctores C. Walton Lillehei y John W. Kirklin llevaron a cabo con éxito operaciones intracardiacas con CEC en algunos pacientes1. Desde aquellas históricas fechas hasta el presente se ha avanzado y perfeccionado de forma muy significativa en el diseño, la fabricación y el desarrollo de las bombas de CEC. Se han incorporado nuevos sistemas de impulsión de la sangre, oxigenadores ultrafinos de membrana y materiales más biocompatibles. Así y todo, la CEC es un procedimiento agresivo que puede provocar complicaciones en muchos órganos y sistemas y, especialmente, inducir el llamado «síndrome de respuesta inflamatoria sistémica». La gran mayoría de los procedimientos quirúrgicos en la cirugía cardiaca se realizan bajo CEC, con pinzamiento aórtico y paro cardiaco selectivo. De esta manera se logra trabajar con un corazón sin movimiento y exangüe. Para prevenir los efectos deletéreos de la isquemia por la falta de perfusión arterial y de oxigenación miocárdica durante el tiempo de pinzamiento aórtico, se usa básicamente solución cardiopléjica hiperpotasémica, con multitud de variantes en cuanto a su composición, intervalos y rutas de administración, dosificación, etc. A pesar del uso de estas soluciones cardiopléjicas y otros métodos de protección miocárdica, un número significativo de pacientes presenta disfunción ventricular e insuficiencia cardiaca post-CEC, en forma de daño miocárdico por isquemia-reperfusión (I-R) o infarto de miocardio (IAM).
En cardiocirugía con CEC y pinzamiento aórtico, un número variable de pacientes pueden sufrir fenómenos de I-R miocárdico o IAM transoperatorio.
La incidencia de IAM perioperatorio en cirugía cardiaca con CEC es variable y puede oscilar entre el 0 y el 29,2%2.
El daño miocárdico por I-R se puede producir cuando se suspende el flujo arterial coronario y posteriormente se reinstaura. La repercusión y la magnitud del daño miocárdico depende, entre otras circunstancias, de la importancia y dominancia de la arteria coronaria afectada, el tiempo de isquemia, el área miocárdica en riesgo y la presencia o ausencia de circulación coronaria colateral.
La revascularización miocárdica, ya sea por trombólisis farmacológica, métodos de cardiología intervencionista coronaria mediante angioplastia/«stent» o cirugía de derivación coronaria (DC), representan los mejores métodos de tratar la isquemia miocárdica; sin embargo, pueden producir el llamado daño o lesión por reperfusión.
Si la reperfusión no se efectúa de forma precoz y efectiva se produce una zona de muerte celular o necrosis3. El daño por I-R puede ser multifactorial4 y produce en el miocito necrosis, apoptosis y autofagia que se pueden presentar simultáneamente. Para muchos investigadores, como García-Dorado et al.3, Piper et al.5, Mc Cully et al.6, Ruiz-Meana et al.7, Kung et al.8, Bonora et al.9 y otros, en el daño miocárdico por I-R la necrosis es el hecho fundamental o único.
En el campo de la cirugía cardiaca el daño por I-R puede aparecer tras una operación de DC con o sin CEC, trasplante cardiaco y en todas las operaciones cardiacas con CEC al despinzar la aorta y reperfundir el miocardio tras un periodo de isquemia.
Durante el pinzamiento aórtico con paro cardiaco selectivo, el miocardio es protegido mediante hipotermia moderada desde la bomba de CEC, hipotermia tópica administrando suero helado en el saco pericárdico y principalmente a través de la administración de cardioplejía. La cardioplejía se puede inyectar anterógada (directamente en los 2 ostium coronarios o bien por la raíz aórtica) o retrógrada por una cánula a través del seno coronario. Puede tratarse de una cardioplejía cristalina o bien hemática, y se puede dar de forma continua o intermitente. Se puede administrar fría, caliente o tibia. La técnica de cardioplejía hemática, fría, intermitente, anterógrada-retrógrada con una inyección de cardioplejía caliente antes del despinzamiento aórtico, es muy utilizada. Actualmente, para muchos cirujanos cardiacos la cardioplejía sigue constituyendo un pilar muy importante en protección miocárdica. A pesar de los avances en los tipos, las modalidades y las técnicas de cardioplejía, un número significativo de pacientes pueden presentar alteraciones miocárdicas en forma de isquemia miocárdica, aturdimiento miocárdico, daño por I-R, necrosis, apoptosis o IAM.
El daño de I-R miocárdico desempeña un papel importante en los enfermos intervenidos de cirugía cardiaca con CEC, siendo el grupo de más edad y con más factores de riesgo los más proclives a presentar complicaciones intraoperatorias y postoperatorias10. Además de la cardioplejía hay otros métodos de protección miocárdica demostrados, como el condicionamiento miocárdico (precondicionamiento isquémico, poscondicionamiento isquémico y poscondicionamiento isquémico remoto) y la cardioprotección con fármacos3,10.
Se presenta un trabajo de revisión de la I-R miocárdica en cirugía cardiaca con CEC, con especial dedicación a los cambios celulares y bioquímicos. Tal y como se define en el trabajo de Castaño-Ruiz11, en el daño miocardio por I-R se pueden contemplar los siguientes efectos: cambios celulares durante la I-R, acción de los radicales libres derivados del oxígeno (RLO), activación del sistema del complemento, activación de las citocinas proinflamatorias e interacción del neutrófilo-endotelio.
Cambios celulares durante la isquemia-reperfusiónEn condiciones normales, la mitocondria del cardiomiocito consume altas concentraciones de O2, genera adenosín trifosfato (ATP) por la vía aeróbica y produce barrenderos de oxígeno para contrarrestar a los RLO; además, también interviene en la homeostasis del Ca12. El Ca del citosol es imprescindible para la contracción del músculo cardiaco, de tal forma que los niveles de Ca citosólico están controlados y regulados de forma precisa13.
Cambios celulares durante la isquemiaLa falta de oxígeno a nivel de la mitocondria afecta a la cadena respiratoria mitocondrial y detiene la síntesis de ATP por la vía aeróbica. El protón Fo F1 ATP-sintasa que normalmente produce ATP cambia a una forma invertida en forma de Fo F1 ATP-asa que consume ATP12. Se comienza a sintetizar ATP por la vía anaeróbica que genera ácido láctico y provoca acidosis celular. El ATP existente se hidroliza por la Na/K-ATPasa y otras enzimas, y como consecuencia aumentan los niveles de adenosín difosfato. Se activa la glucólisis anaeróbica y se generan lactato y protones que inducen una caída del pH celular de 7,4 a 6,4n con acidosis celular y aumento de H+12. La acidificación celular estimula los transportadores sarcolemales alcalinizantes que son el cotransportador Na/HCO3 (NBC) y el intercambiador Na/H (NHE). Estos 2 sistemas antiacidosis celular provocan la entrada en la célula de altas concentraciones de Na, el cual produce disfunción contráctil. El aumento del Na intracelular acelera el intercambiador o bomba Na/K que se activa de forma invertida, lo cual provoca en el miocito una entrada de Ca y salida de Na12. El retículo sarcoplásmico (RSa) no capta Ca del citosol, ya que su transportador necesita ATP, lo que empeora la situación de alto nivel de Ca celular3, lo cual activa las nucleasas, las fosfolipasas y las proteasas provocando destrucción de la membrana celular y muerte celular12.
Cambios celulares durante la reperfusiónLa restauración del flujo coronario hace que se recupere la actividad de la cadena respiratoria, el potencial de membrana mitocondrial y la síntesis de ATP12. Durante los primeros minutos de la reperfusión las bombas o intercambiadores NBC y NHE tienden a normalizar y corregir la acidificación celular. Sin embargo, después de tiempos de isquemia largos o por otras causas, el Na intracelular puede seguir elevado, lo cual activa la bomba Na/Ca en su forma inversa y provoca una entrada de Ca que agrava la sobrecarga de Ca en el citosol12. Los aumentos de Ca citosólico estimulan la captación de Ca por el RSa hasta superar el umbral de los canales de rianedina, con lo cual el nivel de Ca aumenta en el citosol.
Las oscilaciones citosólicas del Ca y sobre todo su aumento provocan una contracción en el cardiomiocito llamado hipercontractura. La hipercontractura es la causante fundamental de la necrosis en bandas de contracción 3. Ultraestructuralmente se puede apreciar rotura sarcolemal, edema mitocondrial, depósito de Ca en la mitocondria y acortamiento y desorganización de las microfibrillas14.
La elevación del Ca citosólico activa sistemas enzimáticas Ca dependientes, como las fosfolipasas, que provocan hidrólisis de los fosfolípidos y acción detergente de proteínas anfipáticas, las cuales producen alteraciones de la membrana celular13.
El aumento de Ca celular durante la reperfusión activa las calpaínas que son unas cistinproteasas no lisosomales y Ca dependientes. Las calpaínas son reguladas e inhibidas por la calpastina. No se sabe con exactitud el papel de las calpaínas en el daño miocárdico por I-R. Algunos trabajos han demostrado que la actividad de las calpaínas provoca la degradación de proteínas estructurales como la fodrina, la desmina y la anquirina. La degradación de fodrina se ha asociado a un aumento de la fragilidad sarcolemal. La degradación de fodrina y anquirina pueden aumentar la sobrecarga de Na y K, y contribuir al daño por reperfusión15.
Durante la reperfusión se observa un secuestro de H2O dentro del miocito y en el espacio extracelular, lo cual produce un edema de miocito y miocardio12.
Las mitocondrias superproducen RLO capaces de peroxidar los ácidos grasos celulares, destruir la membrana mitocondrial y celular, y producir daño por estrés oxidativo.
También la reperfusión conlleva a un aumento de la fragilidad celular12 y a una baja producción de óxido nítrico (ON)3. La baja disponibilidad de ON disminuye la vía de señalización guanosín monofosfato cíclico (GMPc) y proteína cinasa dependiente de GMPc (PKG)3.
La sobrecarga de Ca y RLO inducen la apertura del poro de permeabilidad mitocondrial transicional (PPMT). Si la apertura del PPMT es de duración media o larga, se producen daños irreversibles en el miocito que lo abocan a la muerte celular. En esta situación se produce un colapso en el potencial de membrana mitocondrial y se detiene la síntesis de ATP, hay una depleción del ATP y nicotinamida adenina dinucleótido, liberación de Ca de la mitocondria hacia el citosol, hinchazón de la matriz mitocondrial y rotura externa de la matriz mitocondrial con salida de Ca y otras moléculas de la mitocondria hacia el citosol3,12. Además, la apertura del poro produce la salida hacia el citosol del citocromo C y de las proteínas Smac/DIABLO (Second Mitochondria-derived activator of caspase/Direct IAP Binding protein with Low PI), HTRA2/OMI (high temperature requirement), factor inductor de la apoptosis, endonucleasa G y activator de la caspasa DNase (CAD) que a través de la vía de las caspasas produce la muerte celular por vía de la apoptosis16.
Radicales libres derivados del oxígenoNormalmente, las células en su metabolismo producen RLO pero son rápidamente eliminados por sistemas barrenderos o eliminadores de RLO. Los RLO inorgánicos son el oxígeno molecular, radical superóxido, radical hidroxilo y peróxido de hidrógeno. Los RLO orgánicos son el radical peróxido, hidroperóxido orgánico y lípidos peroxidados. Los sistemas antioxidantes son la catalasa, glutatión-peroxidasa, superóxido-dismutasa, antioxidantes no enzimáticos, proteasas y antioxidantes terciarios. Los llamados barrenderos de los radicales libres (oxygen free radical scavengers) tienen la capacidad de eliminar los RLO y los más importantes son el glutatión, la vitamina C y la vitamina E o tocoferol17.
En la I-R se generan RLO, especialmente en la reperfusión11. Los RLO provocan: 1) peroxidación de los lípidos de la membrana celular del miocito y sus organelas, así como oxidación de las proteínas estructurales y enzimas; 2) inhibición de la fosforilización oxidativa de las mitocondrias; 3) inactivación del ON; 4) estimulación del sistema del complemento; 5) activación de las células endoteliales, y 6) aumento de la expresión del factor de transcripción kappa B (FTK-B)11.
El FTK-B activado induce la transcripción de genes de activación de citocinas proinflamatorias, como las interleucinas (IL) IL-1 beta, IL-2, IL-6, IL-8, factor de necrosis tumoral (TNF-alfa), interferón (INF)-gamma y linfotoxinas. Además activa las moléculas de adhesión ICAM, selectina E y VCAM. También este factor de transcripción interviene en: 1) producción de quimiocinas (quimioatractores celulares que facilitan la infiltración celular) como MIP-1 y IL-8; 2) exposición de proteínas de fase aguda excretadas por los hepatocitos que provocan aumento de los niveles hemáticos de fibrinógeno, amiloide sérico-A, fibrinógeno-beta, metaloproteína-1, alfa-1 antiquimotripsina, alfa-2 macroglobulina, haptoglobina, hemopexina, y otras proteínas relacionadas con el transporte, coagulación y remodelado tisular; 3) exposición de las enzimas ON sintasa (NOS) y ciclooxigenasa-2; 4) formación de factor activador de las plaquetas, y 5) formación de leucotrienos11.
El FTK-B (familia de 5 factores) es una proteína perteneciente a la familia de factores de transcripción nuclear que son proteínas que reconocen secuencias específicas de ADN en la región reguladora de los genes. La unión de estos factores de transcripción a la zona reguladora de los genes modula positiva o negativamente la expresión y subsecuente producción de proteínas codificadas para el gen en cuestión. Normalmente, el FTK-B está en forma inactiva en el citoplasma celular acoplado a la proteína inhibidora factor inhibidor kappa B (IKB) que lo mantiene inactivo e inhibido. Por los pasos de fosforilización, ubiquitinación y degradación de la proteína IKB, el FTK-B se independiza y se transloca al núcleo donde se hace activo uniéndose a secuencias específicas en las regiones promotoras de los genes y activa la transcripción11.
Activación del sistema del complementoEl sistema del complemento está compuesto de unas 30 proteínas del suero que constituyen una cadena enzimática que permite una ampliación de la respuesta humoral. La activación y la fijación del complemento a microorganismos es un importante mecanismo efector del sistema inmunitario que facilita la eliminación del antígeno y genera una respuesta inflamatoria. La activación y la fijación del complemento producen lisis de microorganismos o células diana, opsonización, aumento de la quimiotaxis, aumento de la respuesta humoral y eliminación de los inmunocomplejos. El complemento se puede activar por la vía clásica, la vía alternativa y la vía de las lecitinas. Las 3 rutas comportan la formación del complejo de ataque de membrana (CAM). El complemento genera anafilotoxinas C3a, C3b y C5a que acaban produciendo el complejo citotóxico C5b-9. El complemento normalmente está autorregulado. La CEC dispara la vía del complemento por 3 mecanismos que son el contacto de la sangre con superficies artificiales no endotelizadas, la reperfusión y la administración de protamina11. La CEC solo activa la vía clásica y la alternativa, que acaban produciendo C3, el cual activa el C5 dando lugar a la generación de C3a y C5a, que son anafilotoxinas que estimulan la liberación de histamina y otros mediadores de la inflamación. Las anafilotoxinas producen quimiotaxis de los polimorfonucleares (PMN), estimulación de la actividad inflamatoria y formación de CAM que provocan la destrucción celular13.
Activación de las citocinas proinflamatoriasLas citocinas constituyen un grupo de glucoproteínas de bajo peso molecular (generalmente menor de 30 KDa), producidas durante la respuesta inmunitaria, la inflamación y otras situaciones. Se unen a receptores específicos de la membrana de las células donde van a ejercer su función, iniciando una cascada de transducción intracelular de señal que altera el patrón de expresión génica, de forma que esas células diana producen una determinada respuesta biológica. Las citocinas ejercen su acción al unirse a receptores específicos para cada citocina en la célula en la que ejercen su acción. Existen distintas clases de citocinas con múltiples funciones biológicas. Algunas tienen funciones similares y otras antagónicas. Se pueden considerar los siguientes grupos de citocinas: IL, TNF, INF, factores estimulantes de las colonias y quimiocinas. En la I-R se liberan las citocinas proinflamatorias IL-1, IL-6, IL-8, IL-10, IL-12, TNF-alfa e INF-gamma, las cuales provocan activación de los leucocitos, quimiotaxis de los PMN e infiltración endotelial11.
Interacción neutrófilo-endotelioDurante la CEC y en la I-R miocárdica se activan las células endoteliales y los neutrófilos, que acaban emigrando por diapédesis a través del endotelio e infiltrando la zona de I-R o IAM. Este proceso está dividido en 3 fases10. La fase 1 o de contacto, en la que los neutrófilos disminuyen de velocidad por el vaso y contactan con el endotelio; fase 2 de adhesión mediada por las moléculas de adhesión L-selectina (LAM-1), P-selectina (GMP-140) y E-selectina (ELAM-1), y la fase 3 de migración o diapédesis. Los neutrófilos emigrados y activados activan las proteasas (colagenasa, elastasa, metaloproteasas), RLO, ácido hipocloroso, tromboxanos y leucotrienos; además, inactivan antiproteasas naturales del plasma y medio intersticial11,12.
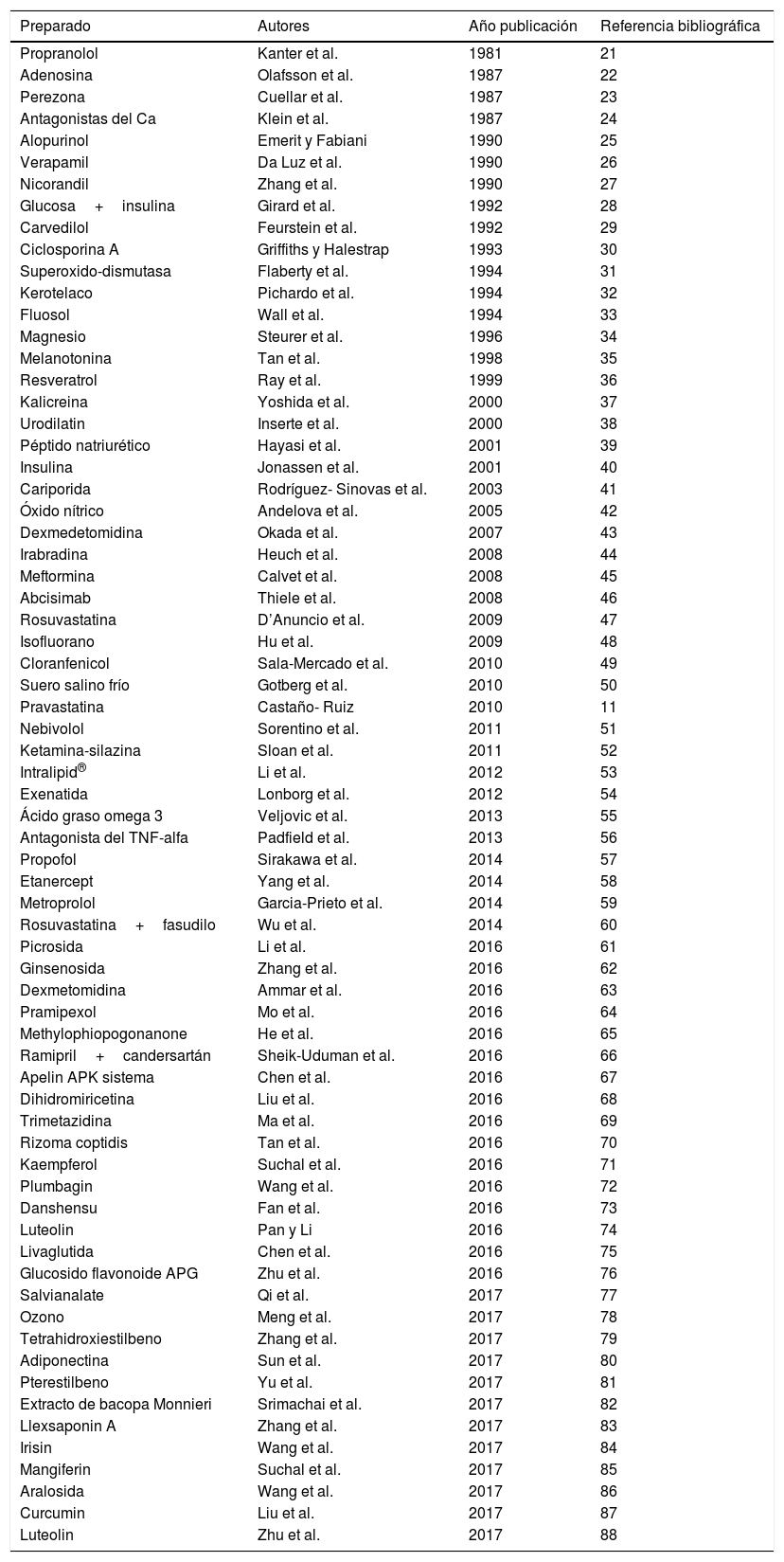
Experimentación con fármacos y compuestos contra el daño de isquemia-reperfusión miocárdicoA lo largo de los años se han utilizado, tanto experimental como clínicamente, muchos compuestos, preparaciones y fármacos para contrarrestar el daño miocárdico por I-R o/y IAM18-20 (tabla 1)11,21-88.
Algunos fármacos, preparados y compuestos usados o ensayados a nivel experimental y clínico contra la isquemia-reperfusión miocárdica y el infarto de miocardio
| Preparado | Autores | Año publicación | Referencia bibliográfica |
|---|---|---|---|
| Propranolol | Kanter et al. | 1981 | 21 |
| Adenosina | Olafsson et al. | 1987 | 22 |
| Perezona | Cuellar et al. | 1987 | 23 |
| Antagonistas del Ca | Klein et al. | 1987 | 24 |
| Alopurinol | Emerit y Fabiani | 1990 | 25 |
| Verapamil | Da Luz et al. | 1990 | 26 |
| Nicorandil | Zhang et al. | 1990 | 27 |
| Glucosa+insulina | Girard et al. | 1992 | 28 |
| Carvedilol | Feurstein et al. | 1992 | 29 |
| Ciclosporina A | Griffiths y Halestrap | 1993 | 30 |
| Superoxido-dismutasa | Flaberty et al. | 1994 | 31 |
| Kerotelaco | Pichardo et al. | 1994 | 32 |
| Fluosol | Wall et al. | 1994 | 33 |
| Magnesio | Steurer et al. | 1996 | 34 |
| Melanotonina | Tan et al. | 1998 | 35 |
| Resveratrol | Ray et al. | 1999 | 36 |
| Kalicreina | Yoshida et al. | 2000 | 37 |
| Urodilatin | Inserte et al. | 2000 | 38 |
| Péptido natriurético | Hayasi et al. | 2001 | 39 |
| Insulina | Jonassen et al. | 2001 | 40 |
| Cariporida | Rodríguez- Sinovas et al. | 2003 | 41 |
| Óxido nítrico | Andelova et al. | 2005 | 42 |
| Dexmedetomidina | Okada et al. | 2007 | 43 |
| Irabradina | Heuch et al. | 2008 | 44 |
| Meftormina | Calvet et al. | 2008 | 45 |
| Abcisimab | Thiele et al. | 2008 | 46 |
| Rosuvastatina | D’Anuncio et al. | 2009 | 47 |
| Isofluorano | Hu et al. | 2009 | 48 |
| Cloranfenicol | Sala-Mercado et al. | 2010 | 49 |
| Suero salino frío | Gotberg et al. | 2010 | 50 |
| Pravastatina | Castaño- Ruiz | 2010 | 11 |
| Nebivolol | Sorentino et al. | 2011 | 51 |
| Ketamina-silazina | Sloan et al. | 2011 | 52 |
| Intralipid® | Li et al. | 2012 | 53 |
| Exenatida | Lonborg et al. | 2012 | 54 |
| Ácido graso omega 3 | Veljovic et al. | 2013 | 55 |
| Antagonista del TNF-alfa | Padfield et al. | 2013 | 56 |
| Propofol | Sirakawa et al. | 2014 | 57 |
| Etanercept | Yang et al. | 2014 | 58 |
| Metroprolol | Garcia-Prieto et al. | 2014 | 59 |
| Rosuvastatina+fasudilo | Wu et al. | 2014 | 60 |
| Picrosida | Li et al. | 2016 | 61 |
| Ginsenosida | Zhang et al. | 2016 | 62 |
| Dexmetomidina | Ammar et al. | 2016 | 63 |
| Pramipexol | Mo et al. | 2016 | 64 |
| Methylophiopogonanone | He et al. | 2016 | 65 |
| Ramipril+candersartán | Sheik-Uduman et al. | 2016 | 66 |
| Apelin APK sistema | Chen et al. | 2016 | 67 |
| Dihidromiricetina | Liu et al. | 2016 | 68 |
| Trimetazidina | Ma et al. | 2016 | 69 |
| Rizoma coptidis | Tan et al. | 2016 | 70 |
| Kaempferol | Suchal et al. | 2016 | 71 |
| Plumbagin | Wang et al. | 2016 | 72 |
| Danshensu | Fan et al. | 2016 | 73 |
| Luteolin | Pan y Li | 2016 | 74 |
| Livaglutida | Chen et al. | 2016 | 75 |
| Glucosido flavonoide APG | Zhu et al. | 2016 | 76 |
| Salvianalate | Qi et al. | 2017 | 77 |
| Ozono | Meng et al. | 2017 | 78 |
| Tetrahidroxiestilbeno | Zhang et al. | 2017 | 79 |
| Adiponectina | Sun et al. | 2017 | 80 |
| Pterestilbeno | Yu et al. | 2017 | 81 |
| Extracto de bacopa Monnieri | Srimachai et al. | 2017 | 82 |
| Llexsaponin A | Zhang et al. | 2017 | 83 |
| Irisin | Wang et al. | 2017 | 84 |
| Mangiferin | Suchal et al. | 2017 | 85 |
| Aralosida | Wang et al. | 2017 | 86 |
| Curcumin | Liu et al. | 2017 | 87 |
| Luteolin | Zhu et al. | 2017 | 88 |
A pesar de que numerosas publicaciones han demostrado la falta de eficacia clínica de muchos productos utilizados, la investigación en cardioprotección miocárdica prosigue activa a nivel de experimentación animal y en estudios clínicos de nuevos fármacos y compuestos dirigidos a minimizar el daño por I-R19. Son necesarios nuevos estudios clínicos, especialmente prospectivos, aleatorizados y doble ciego que demuestren la efectividad de fármacos y preparados contra en daño miocárdico por I-R en cirugía cardiaca con CEC. El cirujano cardiaco debe prestar máxima atención en la protección miocárdica y estar bien informado de nuevas estrategias en cardioprotección.
Conflicto de interesesLos autores declaran no tener conflictos de interés.
Los autores agradecen la colaboración y ayuda en la búsqueda de bibliografía, revisión y asistencia técnica a Carlos C. Abad Torres, estudiante, Colegio Canterbury, Las Palmas de Gran Canaria, España.