




Introducción. A pesar de la frecuencia epidemiológica del carcinoma no medular de tiroides, sobre todo del papilar, son excepcionales los casos descritos de asociación familiar. Presentamos 8 casos de esta enfermedad pertenecientes a 3 familias.
Pacientes y método. De los pacientes intervenidos en nuestro hospital por carcinoma no medular, se seleccionaron aquellos en los que existía asociación familiar sin relación con los síndromes de Gardner, de Coluden ni el MEN-1 y no habrán tenido exposición previa a la radiación.
Resultados. La edad media de los pacientes fue de 46 años, siendo 7 mujeres (87,5%). Excepto en uno de los casos, que consultó por hipercalcemias y fue intervenido con el diagnóstico de hiperparatiroidismo primario, en los demás la palpación cervical y la ecografía demostraban la presencia de un nódulo tiroideo. En estos 7 casos se realizó punción del nódulo, que en seis de ellos fue indicativa de carcinoma papilar. En todos se realizó tiroidectomía total, y en 4 casos en los que se objetivaron adenopatías, ésta se completó con un vaciamiento ganglionar funcional. Todos los carcinomas papilares eran de la variedad bien diferenciado, con un tamaño medio de 1,5 ± 0,9 cm, siendo en 4 casos (50%) un microcarcinoma. Tres pacientes presentaron adenopatías positivas (37,5%). Todos fueron tratados posquirúrgicamente con yodo-131, excepto la paciente con hiperparatiroidismo, que presentaba un microcarcinoma de 0,5 cm. Con un seguimiento medio de 2,5 años, los controles son normales, excepto en un caso, en el que se ha detectado una tiroglobulina elevada, con exploración clínica y rastreo morfológico normales, a los 7 años de la cirugía, por lo que ha recibido tratamiento con 100 mCi de yodo-131.
Conclusiones. El carcinoma no medular familiar de tiroides es una enfermedad inusual, con frecuente afectación ganglionar en el momento del diagnóstico, que presenta un relativo buen pronóstico con un tratamiento acorde con el estadio de la enfermedad.
Introduction. Despite the incidence, in epidemiological terms, of nonmedullary thyroid carcinoma, especially the papillary type, a familial association is rarely described. We present eight cases of this disease occurring in three families.
Patients and method. Among the patients treated surgically for nonnodular carcinoma in our hospital, we selected those showing a familial relationship, in whom the Garden syndrome, the Cawden syndrome and multiple endocrine neoplasia type 1 (MEN-1) had been ruled out and who, in addition, had had no previous exposure to radiation.
Results. The mean patient age was 46 years; 7 of the patients (87.5%) were women. One patient presented with hypercalcemia and was treated surgically for primary hyperparathyroidism. In the remainder, palpation and ultrasound revealed the presence of a thyroid nodule which, when examined by needle biopsy, was considered suggestive of papillary carcinoma in six cases. These seven patients underwent total thyroidectomy, accompanied by lymphadenectomy in the four patients in whom there was evidence of lymph node involvement. All the papillary carcinomas were well differentiated lesions with a mean size of 1.5 ± 0.9 cm. There were four cases (50%) of microcarcinoma. Lymph node involvement was confirmed in three patients (37.5%). All the patients were treated postoperatively with iodine-131, with the exception of the one with hyperparathyroidism who presented a microcarcinoma measuring 0.5 cm. After a mean follow-up of 2.5 years, there is no evidence of disease except in one patient who, seven years after surgery, has been found to present an elevated thyroglobulin level with no abnormal clinical or morphological signs, and has been treated with 100 mCi iodine-131.
Conclusions. Familial nonmedullary thyroid carcinoma is an unusual lesion that is frequently associated with lymph node involvement at diagnosis. The prognosis is relatively good when the patient receives the proper treatment in agreement with the disease stage.
Introducción
En la enfermedad neoplásica tiroidea es bien conocida la asociación familiar del carcinoma medular tiroideo en aproximadamente el 20% de los casos, generalmente como MEN-2, o bien como una forma familiar, sin neoplasia en otros tejidos1,2. Sin embargo, la mayoría de las formas de cáncer tiroideo no medular (papilar, folicular y anaplásico) habían sido consideradas generalmente como de aparición esporádica.
El carcinoma papilar ha sido descrito con poca frecuencia en familias1,3, y en los casos comunicados generalmente iba asociado a los síndromes de Gardner4,5, de Cowden5, o MEN-16, a la exposición a radiación en cuello o cabeza7 o en gemelos monocigóticos8, siendo su incidencia familiar al margen de estas situaciones infrecuente7,9-12.
Presentamos 8 casos de carcinoma papilar de tiroides pertenecientes a 3 familias, sin asociación a ninguna de las situaciones mencionadas con anterioridad.
Pacientes y método
En nuestro servicio de cirugía se han intervenido 261 carcinomas papilares de tiroides entre 1970 y 1999. De éstos hemos seleccionado a 8 pacientes (3,1%) correspondientes a tres familias, en los cuales se detectó la existencia de una relación fa miliar.
Antes de su catalogación como carcinoma papilar familiar se descartó la existencia de cuadros clínicos que justificaran la asociación familiar del cuadro:
1. Síndrome de Gardner mediante endoscopia, tránsito gastrointestinal y rastreo óseo4.
2. Síndrome de Cowden mediante endoscopia, ecografía abdominal y mamografía (estas dos últimas en las mujeres)5.
3. Síndrome MEN_16. Ante un hiperparatiroidismo está justificada la realización de estudios de detección de extensión de este síndrome si cumple alguno de los criterios de alta probabilidad de MEN_1 (paciente menor de 40 años, afectación multiglandular de las paratiroides, existencia de hiperplasia de las paratiroides o hiperparatiroidismo recurrente tras la cirugía curativa)6. Al no cumplir nuestro paciente afectado de hiperparatiroidismo ninguno de estos criterios no estaba indicado realizar estudios de detección para este síndrome.
4. Exposición previa a radiación. No había antecedentes en estas familias.
Los pacientes intervenidos con carcinoma papilar familiar presentan el mismo control postoperatorio que el resto de carcinomas papilares intervenidos: a las 4-6 semanas se les realiza una determinación de hormonas tiroideas, tiroglobulina y una gammagrafía de rastreo y, según los restos posquirúrgicos encontrados, se administra una dosis de yodo-131 entre 100 y 200 mCi y se realiza tratamiento sustitutivo con tiroxina (T4) a dosis supresivas de TSH; a los siguientes 4-6 meses se realiza una determinación de hormonas tiroideas, una exploración clínica (si es positiva se realiza ecografía y punción-aspiración con aguja fina de la lesión y se indica tratamiento con yodo-131) y una determinación de tiroglobulina (normal < 2 ng/ml); posteriormente se realiza un rastreo gammagráfico anual (se actúa según hallazgos) y una determinación analítica semestral (tiroglobulina).
Se sometió a los familiares a una exploración clínica, ecografía cervical y determinación de concentraciones de tiroglobulina como prueba de detección, siendo posteriormente seguidos de manera periódica, cada 2 años, en consultas externas, con exploración clínica y tiroglobulina de control.
Resultados
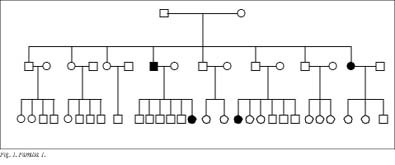
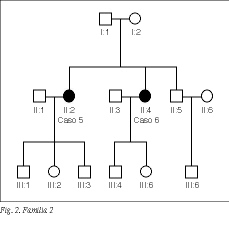
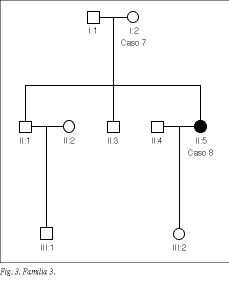
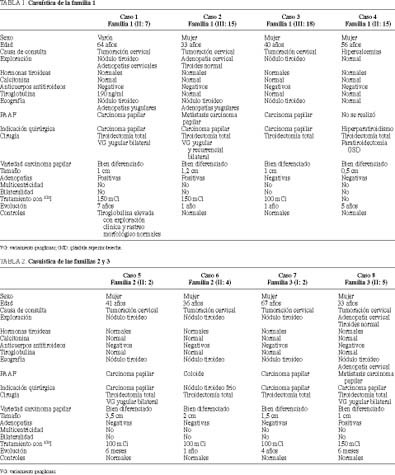
Se han estudiado 8 pacientes afectados de carcinoma papilar familiar, pertenecientes 3 familias (figs. 1-3 y tablas 1 y 2). La edad media fue de 46,3 ± 13,9 años (rango, 33-67 años), siendo 7 casos mujeres (87,5%).




Los pacientes consultaron en 7 casos (87,5%) por tumoración cervical y en uno (12,5%) por clínica derivada de su hipercalcemia. Excepto en el caso del hiperparatiroidismo, la palpación cervical y la ecografía fueron patológicas. Las hormonas tiroideas, la calcitonina y la tiroglobulina presentaron cifras dentro de los límites de la normalidad, excepto un paciente con cifras de tiroglobulina de 190 ng/ml. Excepto en el caso del hiperparatiroidismo, se realizó punción-aspiración con aguja fina, que en 6 casos (85,7%) puso de manifiesto la presencia de un carcinoma papilar.
Los 8 pacientes fueron intervenidos y se les realizó tiroidectomía total. El vaciamiento ganglionar se hizo en los 4 casos en los que se objetivaron adenopatías. En la paciente con hiperparatiroidismo se halló intraoperatoriamente un nódulo de 1 cm de diámetro en hemitiroides derecho, informando la biopsia intraoperatoria de carcinoma papilar, por lo que se realizó tiroidectomía total.
La anatomía patológica confirmó la presencia de carcinoma papilar de la variedad bien diferenciado en todos los casos, con un tamaño medio de 1,5 ± 0,9 cm. En 3 casos se presentaron adenopatías positivas. Todos fueron tratados posquirúrgicamente con yodo-131, excepto la paciente con hiperparatiroidismo, pues presentaba un microcarcinoma papilar tiroideo de 0,5 cm de diámetro sin extensión y con rastreo posquirúrgico normal.
Con un seguimiento medio de 2,5 ± 2,5 años, los controles periódicos posquirúrgicos han sido normales, excepto en un caso, en el que se ha detectado una tiroglobulina elevada, con exploración clínica y rastreo morfológico normales a los 7 años de la cirugía, por lo que recibió tratamiento con yodo-131 (100 mCi).
Discusión
La incidencia familiar de carcinoma no medular de tiroides, en sus tres variedades (papilar, folicular y anaplásico) es extremadamente rara3,13,14. Respecto a los no familiares, los carcinomas papilares familiares se caracterizan por1: a) inicio a una edad más temprana que el esporádico, y b) transmisión vertical en generaciones consecutivas y manifestación horizontal en hermanos: herencia autosómica dominante con expresión variable.
Debido a la incidencia familiar se hace necesaria la realización de un seguimiento para la detección y el tratamiento precoz de los individuos afectados. Lo importante sería el diagnóstico preclínico, por ejemplo genético, hecho que actualmente todavía no es posible15,16. Cuando dos o más pacientes de una misma familia tienen carcinoma papilar de tiroides, todos los de la primera y segunda generación relacionados deberían someterse a un examen del cuello mediante palpación y ecografía, como se realizó en nuestros pacientes. Hay que recordar que la ecografía como prueba de detección presenta una alta sensibilidad con una baja especificidad17.
Los errores genéticos promotores del carcinoma familiar papilar no están claramente establecidos, aunque hay varios estudios que detectan una herencia autosómica dominante con penetrancia reducida10,11. Se ha asociado al HLA; así, Ozaki11 comunicó la presencia del HLA B7 y DR1 significativamente más frecuente en pacientes con carcinoma papilar familiar de tiroides que en los esporádicos; además, el haplotipo B7 Cw7 DR1 se observó en 5 de sus 13 casos. Los cuatro oncogenes que en teoría pueden estar activados en la carcinogénesis tiroidea son el RAS, TRK (tropomyosin receptor kinase), PTC (papillary thyroid carcinoma) (variable del RET) y el RET18-21, de los cuales sólo es reproducible el RET, y ya hay algunos estudios que lo excluyen como causante de la enfermedad3.
Los escasos casos descritos en la bibliografía presentan, en cuanto a la histología una mayor multicentricidad y bilatera lidad que en los casos esporádicos (el 50% frente al 21-65%), mayor enfermedad extratiroidea o invasión local en el mo mento del diagnóstico (el 32 frente al 4-16%), aunque no hay más incidencia de metástasis a distancia en el diagnósti co3,10,11,13,22,23. En nuestros casos, sin embargo, no se objetivó multicentricidad ni bilateralidad, aunque sí había presencia de invasión ganglionar en 3 de los 8 casos.
Las familias con dos o más individuos con carcinoma papilar tiroideo deben ser controlados por dos razones. Una para detectar la presencia de dicho carcinoma, y otra porque hay autores que han notificado un incremento de otras neoplasias malignas, sobre todo de origen gastrointestinal y renal9,12-14,18, en estos pacientes. En nuestros pacientes y sus familiares directos no se ha detectado ninguna neoplasia extratiroidea.
Aunque varios autores recomiendan un tratamiento más agresivo9,12-14,18, con tiroidectomía total más linfadenectomía como cirugía inicial (por la alta incidencia de multifocalidad y bilateralidad), y radioyodo postoperatorio ablativo (para disminuir la incidencia de recidiva locorregional), no se pueden extraer conclusiones según los estudios publicados, para recomendar un tratamiento más agresivo en el subgrupo familiar que no sea el tratamiento acorde al estado de la enfermedad y otros factores de riesgo, como en el carcinoma esporádico. En nuestra serie, el tratamiento fue adecuado al estado de la enfermedad, de forma que se realizó vaciamiento ganglionar cuando había ganglios afectados, y en el caso del microcarcinoma sólo se realizó tiroidectomía, con buenos resultados en los seguimientos realizados en consultas externas.
El cáncer de tiroides diferenciado es una neoplasia con relativo buen pronóstico, por lo que precisa un seguimiento de varios años para su valoración. Así, el caso 1 de nuestra serie (tabla 1) puede presentar una recidiva una vez pasados más de 7 años tras la cirugía, a pesar de presentar afectación ganglionar cuando fue intervenido. Actualmente, el pronóstico en el carcinoma papilar familiar es incierto respecto al esporádico, debido a los datos clínicos limitados de los que se dispone3.
En conclusión, podemos decir que el carcinoma papilar familiar de tiroides es una enfermedad inusual, con frecuente afectación ganglionar en el momento del diagnóstico, que presenta un relativo buen pronóstico con un tratamiento acorde con el estadio de la enfermedad.

