




Sr. Director:
El ganglioneuroma es un infrecuente tumor derivado de las células ganglionares simpáticas, que a su vez se originan embriológicamente en la cresta neural1,2. Representa el miembro más diferenciado de la familia de tumores derivados del sistema nervioso simpático1,3, siendo considerado por muchos autores como el equivalente benigno del neuroblastoma1,4. A diferencia de éste, maligno, con frecuencia irresecable y típico de la infancia, el pronóstico del ganglioneuroma es excelente, su exéresis raras veces no es posible, y su incidencia se eleva a partir de la segunda década de la vida3,5. La localización más frecuente es el mediastino posterior, seguida del retroperitoneo3,6,7; a este nivel asentaba el caso que aquí aportamos.
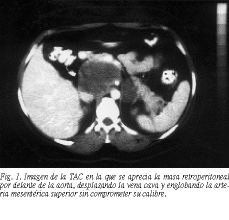
Se trataba de una mujer de 34 años con antecedente de traumatismo abdominal 12 años atrás, a raíz del cual se le detectó una tumoración por detrás de la cabeza del páncreas que fue interpretada como seudoquiste. Unos meses previos a su visita, la paciente inició un cuadro de dolor en costado derecho, pesadez posprandial y náuseas, motivos por los que consultó. La exploración física y constantes vitales fueron normales, así como la radiografía de tórax y abdomen y las analíticas de sangre y orina. Se completó el estudio con ecografía y TAC abdominal (fig. 1) encontrándose una masa retroperitoneal de 10 * 5 cm y baja densidad, por delante de cava y aorta y por detrás de los vasos esplénicos y proceso uncinado del páncreas, polilobulada, rodeando la arteria mesentérica superior sin disminuir su calibre. Se realizó punción-aspiración que no fue diagnóstica, por lo que se decidió la exploración quirúrgica. Tras laparotomía transversa y maniobra de Kocher amplia se accedió a una tumoración blanquecina de consistencia elástica, en íntima relación con las venas cava inferior y renal derecha, aorta y caras posteriores de la arteria hepática, vena porta y páncreas, que fue extirpada en su totalidad. El diagnóstico histológico de la pieza fue de ganglioneuroma. No hubo incidencias en el postoperatorio y la paciente permanece asintomática tras 18 meses de seguimiento.

Para buscar el origen del ganglioneuroma nos debemos remontar a las células más primitivas de la cresta neural, las simpatogonias. Éstas se diferencian en su desarrollo embrionario en dos estirpes celulares: la de tipo feocromocítico, que dará lugar a la médula adrenal, y la de tipo simpatoblástico, que formará el sistema nervioso simpático8. Los tumores derivados de esta segunda estirpe pueden ser clasificados según su grado de malignidad en: a) neuroblastoma, muy maligno; b) ganglioneuroblastoma, de malignidad intermedia, y c) ganglioneuroma, tumor benigno8,9. Los tres tumores están bien definidos patológicamente, pero no representan sino el espectro de maduración y diferenciación de una misma estirpe histológica1,10.
El ganglioneuroma constituye el miembro más diferenciado de la familia1,3,7,10 y está compuesto histológicamente por células ganglionares maduras sobre un estroma neurofibrilar en el que también se observan células de Schwann1,2,5,10,11. La presencia creciente en el tumor de acumulaciones de elementos neuronales menos diferenciados es lo que define al ganglioneuroblastoma en primer término, y finalmente al neuroblastoma1,5,7. Es un hecho comprobado, y en la bibliografía existen múltiples ejemplos, que los neuroblastomas pueden madurar a ganglioneuromas1,4,7; esta maduración se ha descrito tanto de forma espontánea como inducida por tratamiento quimio o radioterápico, y puede producirse tanto en el tumor primario como en sus metástasis1,4,12.
El ganglioneuroma es típico de adultos jóvenes, diagnosticándose entre un 40 y un 60% antes de los 20 años y más del 80% antes de los 402,6,13,14. Parece existir un cierto predominio en el sexo femenino5. Su localización más frecuente es el mediastino posterior seguida del retroperitoneo3,6,7, pero pueden aparecer en cualquier localización que alcance el sistema nervioso simpático15. Se ha descrito su asociación con la neurofibromatosis de Von Recklinghausen7,14-17 y con el síndrome de neoplasias endocrinas múltiples 2b14,15,17; en estas enfermedades es típica la localización gastrointestinal de los ganglioneuromas14,15,17.
En muchas ocasiones, el ganglioneuroma cursa de forma asintomática y su diagnóstico es casual9,16,18. Cuando provoca sintomatología, ésta suele ser el resultado del tamaño y compresión de estructuras vecinas y, por tanto, varía según la localización2-4. Se han descrito síntomas neurológicos por compresión medular si el tumor crece a través de las raíces nerviosas y se introduce en el canal espinal, isquémicos por compromiso vascular, respiratorios, urinarios, digestivos, etc.4,18. También se han publicado casos de diarrea severa asociada a la producción por el tumor de péptido intestinal vasoactivo3,5, si bien éste es un hecho muy poco frecuente. Aunque la presencia de síntomas atribuibles a un exceso de catecolaminas es excepcional, no lo es tanto que los valores en orina de ácido vanilmandélico y homovanílico aparezcan discretamente elevados, en especial en tumores de gran tamaño5. Valores muy elevados de estos metabolitos deben alertarnos sobre posibles focos de neuroblastoma en el tumor5.
La orientación diagnóstica se basa en técnicas de imagen, ecografía, TAC, resonancia magnética o gammagrafía con metaiodobencilguanidina marcada, aunque ninguna de ellas puede confirmar inequívocamente el diagnóstico de ganglioneuroma frente a otros tumores de extirpe neural6,13. En la TAC, la presencia de una masa bien definida, ovoide o polilobulada, con un coeficiente de atenuación bajo o intermedio y escaso realce tras la administración de contraste, sugiere el diagnóstico de ganglioneuroma6,13, más aún si se objetiva la presencia de calcificaciones puntiformes, presentes en un 20% de los casos6. También se ha apuntado como un hallazgo orientativo la tendencia del tumor a rodear total o parcialmente vasos sanguíneos, sin que en la mayoría de los casos estenose su luz13. Esta característica pudimos confirmarla en el caso que presentamos, pues el tumor rodeaba totalmente la arteria mesentérica superior pero no comprometía su calibre. En la resonancia magnética, el tumor es homogéneo, con una relativa baja señal de intensidad en T1, y heterogéneo con una predominante alta señal de intensidad en T2. Este último dato parece ser especialmente característico6,13.
El diagnóstico inequívoco precisa la confirmación histológica, bien en la pieza de resección, bien en las biopsias. Se debe destacar, no obstante, que aproximadamente un 25% de ganglioneuromas contienen componentes pobremente diferenciados, ya sea de ganglioneuroblastoma, de neuroblastoma o incluso de feocromocitoma6,13, por lo que un diagnóstico basado en biopsias sólo puede ser de presunción5. Esta podría ser la causa de que tumores diagnosticados como ganglioneuromas en múltiples biopsias desarrollen metástasis en su evolución4.
En cuanto al tratamiento, éste es esencialmente quirúrgico. Si el diagnóstico de ganglioneuroma se realiza de manera preoperatoria según las biopsias, hay autores que abogan por la abstención terapéutica y realizan únicamente un seguimiento del paciente, con nuevas biopsias si el tamaño tumoral se incrementa, para descartar la aparición de neuroblastoma4. Sin embargo, creemos que esta opción debe considerarse con cautela y únicamente en centros con experiencia en estos tumores pues, como ya hemos comentado, las biopsias sólo aportan un diagnóstico de presunción, la coexistencia de ganglioneuroma con ganglioneuroblastoma, neuroblastoma e incluso feocromocitoma es un hecho comprobado, y ganglioneuromas observados durante años han acabado desarrollando tumores agresivos4,12,13.

