




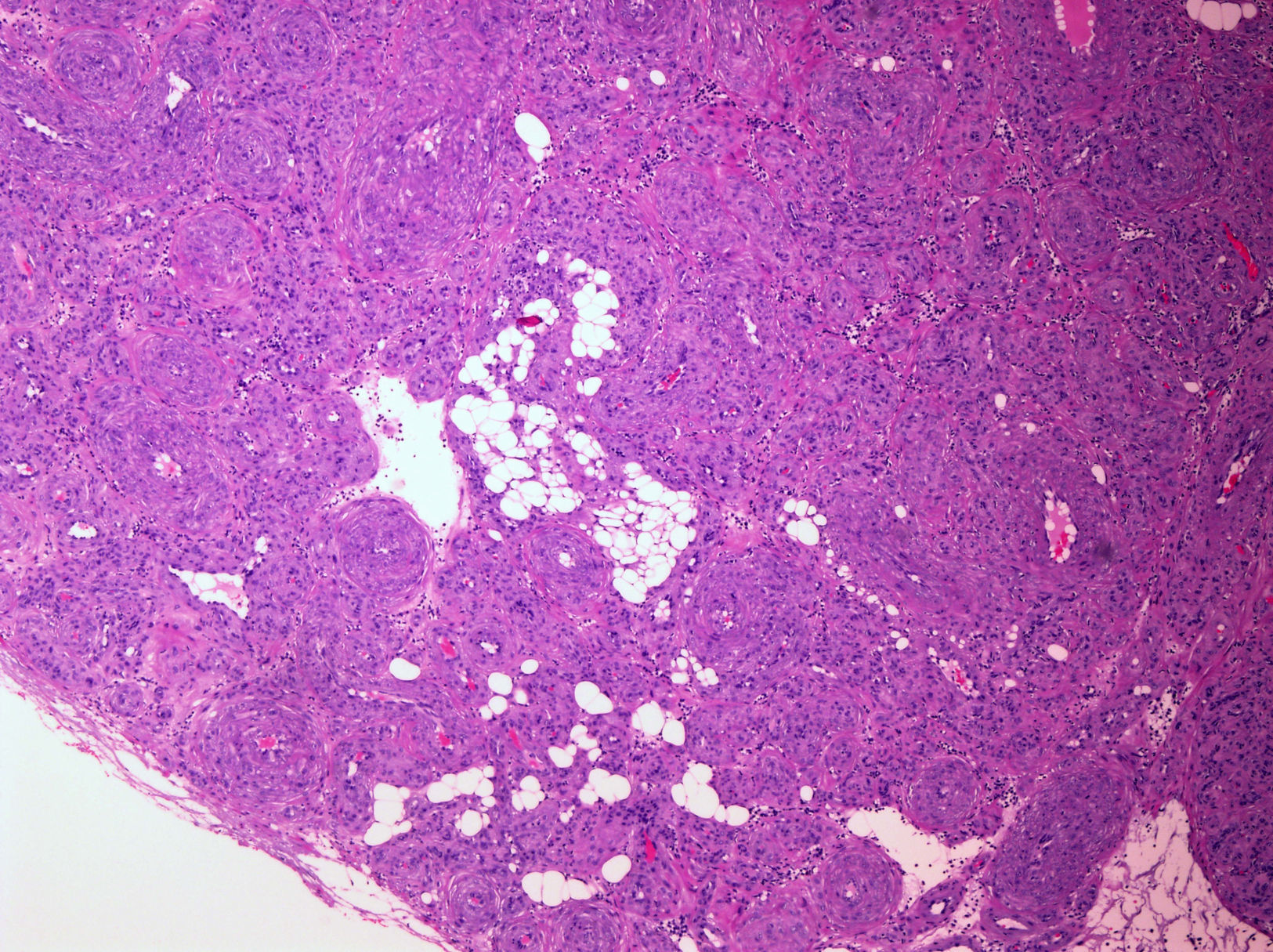
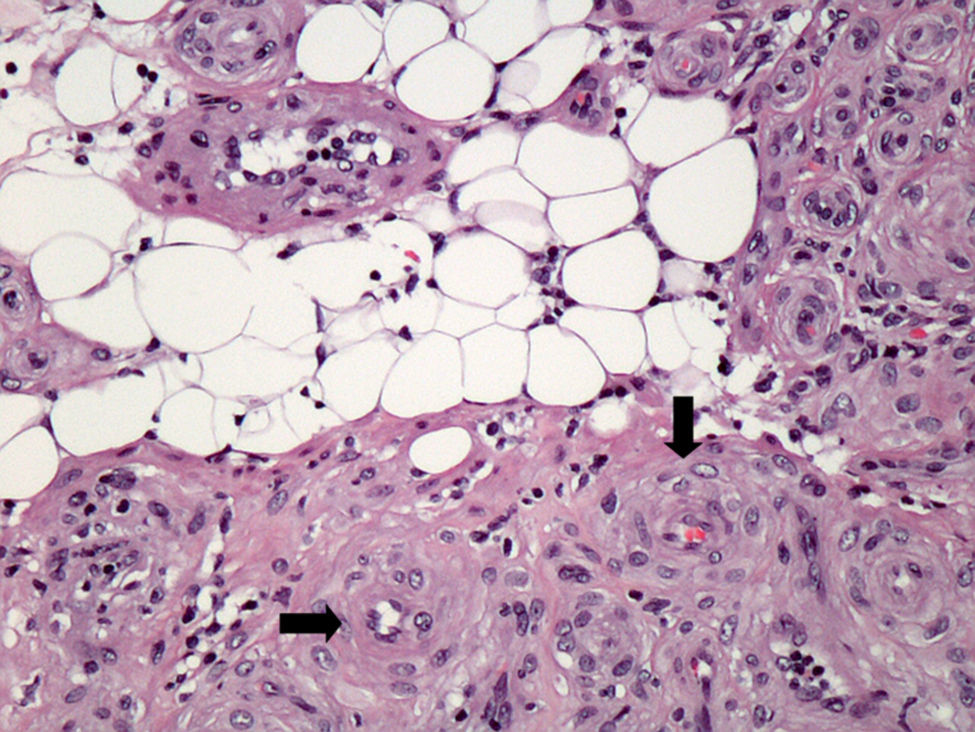
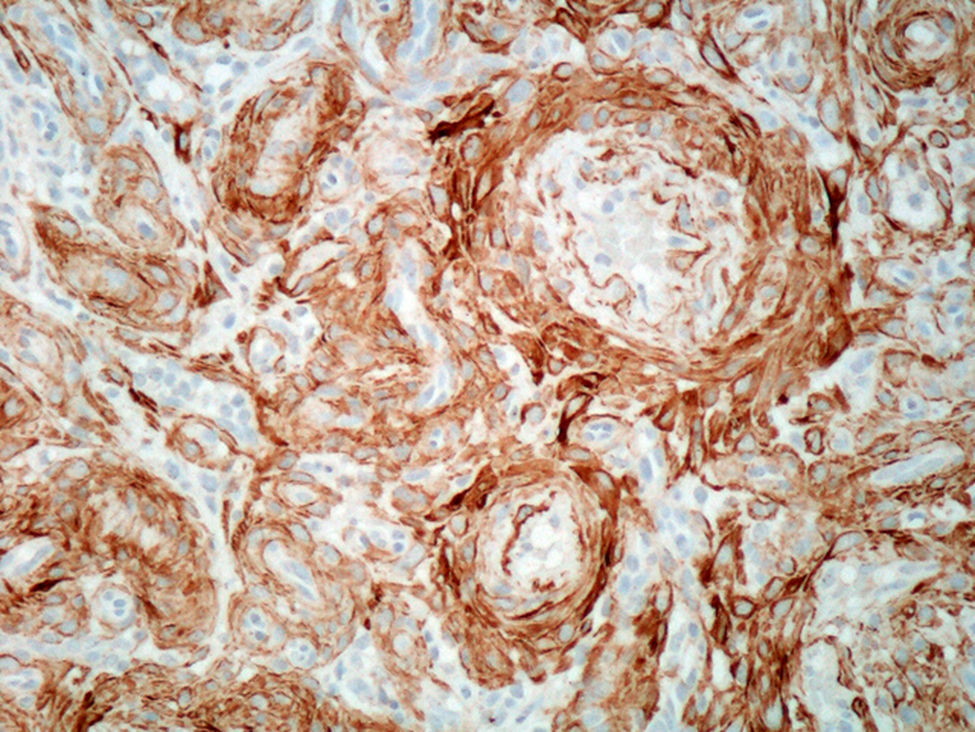
Presentamos un caso de una tumoración nodular subcutánea dolorosa pretibial, vascularizada, de 2,5cm, en una mujer de 82 años de edad. El tumor, con un tiempo de evolución de 21 años, mostró aumento de su tamaño en los últimos 5 años y en un seguimiento a los 4 años de la resección no había recidivado. Tras la extirpación quirúrgica se observó que la lesión no estaba encapsulada pero era bien delimitada. Microscópicamente destacaba una proliferación de elementos vasculares en general de pequeño tamaño, con esporádicos vasos arteriales y venosos de tamaño intermedio (fig. 1), con la particularidad de tener un componente de tejido adiposo maduro en el centro del nódulo (fig. 2, centro). Era muy llamativo un predominio de células fusiformes de pequeño tamaño, núcleo oval, sin atipia, de aspecto mioide con disposición perivascular concéntrica (fig. 2, flechas). No se observaban figuras de mitosis. El estudio de expresión inmunofenotípica mostró positividad difusa perivascular para actina de músculo liso (fig. 3), focal expresión de receptores androgénicos y negatividad en la proliferación perivascular para desmina, CD10, CD34, CD31, S100 (a excepción de las áreas adiposas), HMB45 y melan A. A pesar de la ausencia de mitosis, la tinción para Ki67 mostró hasta 20 células positivas por campo de gran aumento. La morfología de esta lesión planteó inicialmente un diagnóstico diferencial entre tumor fibroso solitario con un componente lipomatoso, angiomiolipoma y angioleiomioma. No obstante, el típico patrón de crecimiento concéntrico perivascular junto con el inmunofenotipo de positividad para actina de músculo liso, negatividad para desmina, HMB45 y melan A nos llevaron al diagnóstico de miopericitoma.
.")
 y presencia de componente de tejido adiposo maduro. (H&E x200).")
.")
Los tumores pericíticos (de crecimiento perivascular) se han denominado tradicionalmente hemangiopericitomas o miofibromas solitarios. Requena et al1 en el año 1996 fueron los primeros en proponer la denominación de miopericitoma (MP) al miofibroma cutáneo del adulto y posteriormente Granter et al2 en el año 1998 adoptaron esta misma terminología, y con este nombre figura en la clasificación de la OMS del año 2002 junto con el tumor glómico y el hemangipericitoma sinonasal, dentro del grupo de los tumores derivados de células perivasculares o pericíticas3. El MP es un tumor benigno, en general dérmico o subcutáneo, nodular, solitario, aunque se han descrito formas multifocales, no doloroso, bien delimitado, menor de 2cm, de crecimiento lento durante años, más frecuente en adultos de edad media y casi siempre localizado en piernas. Esta neoplasia está compuesta por células de aspecto mioide, sin atipia ni actividad mitótica y, típicamente, con gran tendencia a la disposición perivascular concéntrica. Las células tumorales muestran expresión inmunohistoquímica de actina de músculo liso y h-caldesmón y son usualmente negativas para desmina4.
El diagnóstico diferencial del MP se debe establecer fundamentalmente con el angioleiomioma y con el tumor glómico. Existe una variante maligna de MP5 y otra variante intravascular6. Se conoce una variante lipomatosa de hemangiopericitoma, probablemente relacionada con el tumor fibroso solitario7, pero no se ha comunicado ninguna variante lipomatosa de MP.
Los MP usualmente se comportan como tumores benignos, pero en algunos casos puede haber recidiva local, en general relacionada con mala delimitación del tumor y raramente ocurren metástasis en tumores atípicos y malignos4. Respecto al origen de esta neoplasia, se han descrito algunos casos asociados a traumatismo previo8.
La presencia de un componente lipomatoso benigno en nuestro caso, no referido previamente en esta entidad, nos hace proponer para este tumor el término de «miopericitoma lipomatoso», que en su forma de presentación habitual puede plantear diversos diagnósticos diferenciales y creemos que es interesante conocer por parte de los especialistas dedicados a cirugía.

