




El síndrome de Ehlers-Danlos (EDS) es un grupo de trastornos hereditarios poco frecuente (1:5.000 nacidos vivos), que se caracterizan por hiperextensibilidad cutánea, hiperlaxitud articular y fragilidad del tejido conectivo. Dentro de las formas de presentación, la rotura aneurismática es rara, siendo característica en la variante vascular, y excepcional en las demás1. Presentamos un caso de una mujer de 38 años diagnosticada de un EDS tipo clásico, a raíz de un hemoperitoneo secundario a la rotura de un aneurisma gigante de arteria esplénica.
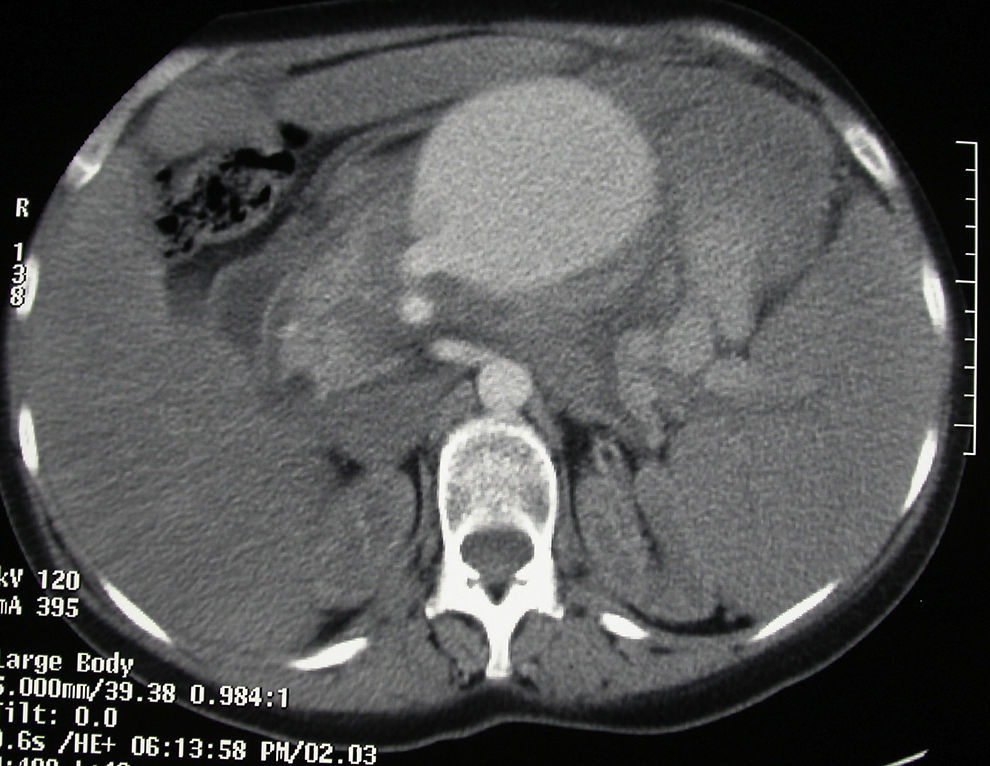
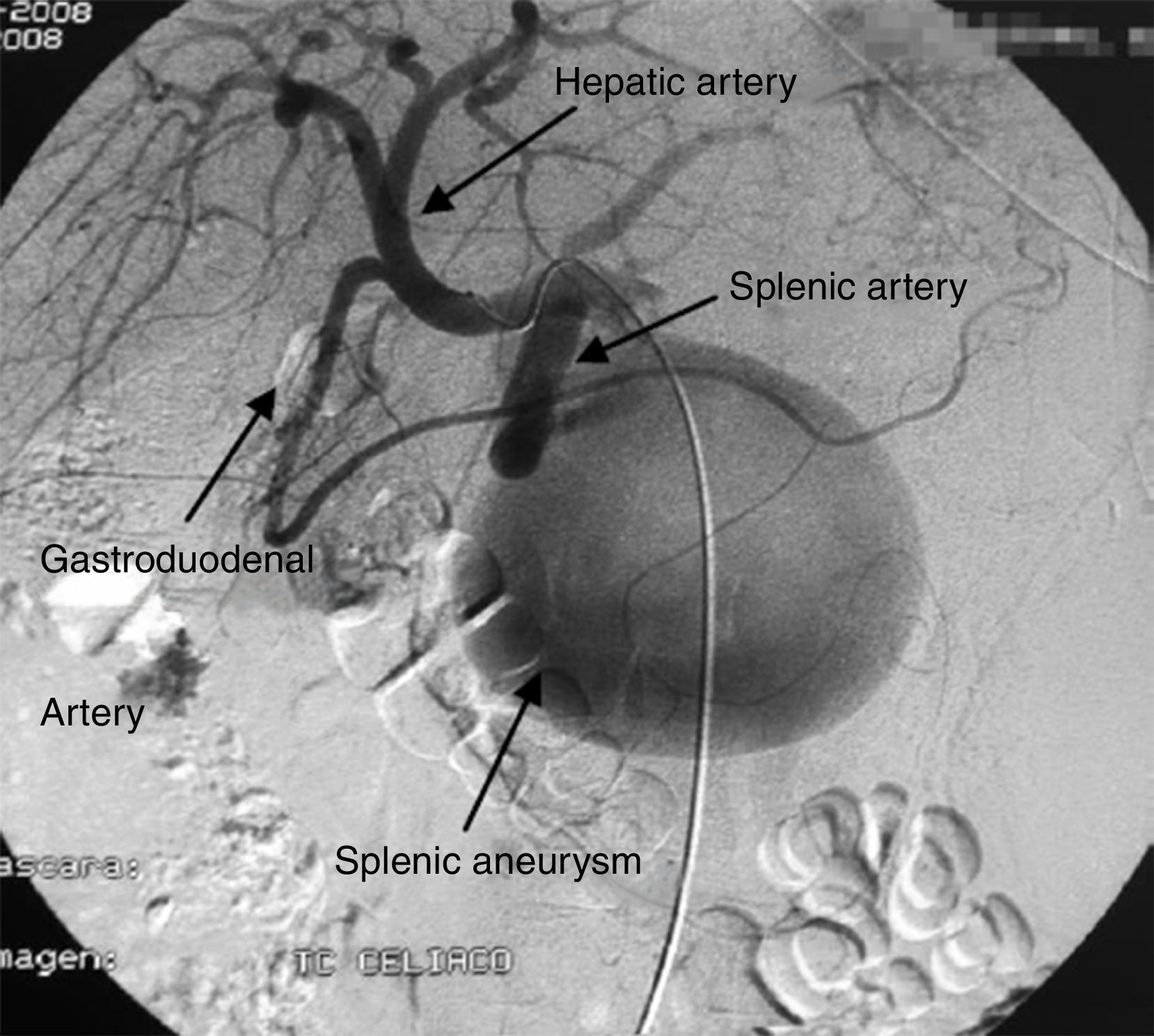
La paciente consultó en urgencias por dolor abdominal y vómitos, no asociado a ningún traumatismo previo. Como antecedentes personales destacaba insuficiencia renal e hipotiroidismo autoinmune. A su llegada a urgencias estaba hemodinámicamente inestable. A la exploración física presentaba abdomen distendido y doloroso a la palpación, asociado a esplenomegalia. En la analítica destacaba un hemograma con hematocrito 10% y hemoglobulina 3,3g/dl. Se realizó una TC abdominal (fig. 1) y una angiografía (fig. 2), donde se objetivó a 4cm del origen de la arteria esplénica un gran aneurisma y 2 seudoaneurismas localizados en hilio esplénico. Se realizó embolización del mayor, con trombina y coils metálicos. A pesar del tratamiento intervencionista, la paciente empeoró hemodinámicamente, por lo que se decidió intervenir hallando hemoperitoneo de más de 2l con esplenomegalia (aproximadamente de 30cm) y seudoaneurisma sobre cuerpo de páncreas parcialmente roto en polo inferior de aproximadamente 10cm. Se realizó disección hiliar con ligadura transfixiva de esplénica y esplenectomía reglada. La paciente evolucionó favorablemente siendo alta al décimo día del postoperatorio.


La paciente presentaba un aspecto leptosómico y fenotipo marfanoide, por lo que se derivó a la consulta de genética médica, y al finalizar el estudio se detectó una mutación en el gen COL5A1 (c.1588G>A) que a nivel de la proteína produce el cambio de la glicina de la posición 530 por una serina (p.Gly530ser).
El EDS se clasifica según Villefranche en 6 subtipos principales. El tipo clásico, el tipo asociado a hipermovilidad y el tipo vascular son los más comunes, mientras que la cifoescoliosis, arthrochalasis y el tipo dermatosparaxis constituyen condiciones muy raras. La mayoría de las formas de EDS corresponden a mutaciones en genes que codifican cadenas de colágeno o enzimas implicadas en la biosíntesis de estas proteínas1. En el tipo clásico, alrededor del 50% de los casos, el defecto se encuentra causado por mutaciones en los genes que codifican el colágeno V alfa-1 (COL5A1) o alfa-2 (COL5A2)2,3. Ante la sospecha de un EDS debemos buscar antecedentes que incluyan complicaciones, tales como hematomas frecuentes, insuficiencia cervical, prolapso anal en la infancia, rotura prematura de membranas, laceraciones vaginales, baja capacidad pulmonar o soplos3,4. La frecuencia y severidad de los síntomas digestivos en el EDS depende del tipo, siendo la forma vascular la que presenta mayores complicaciones gastrointestinales. Existe escasa bibliografía sobre casos de rotura hepática, biliar o esplénica espontánea, probablemente debido a una menor cantidad de tejido de soporte estructuralmente intacta en estos órganos5.
En el diagnóstico diferencial de dolor abdominal agudo en el EDS, el sistema vascular siempre debe ser considerado. La mayoría de los subtipos de EDS tienen diátesis hemorrágica, a pesar de perfiles de coagulación normales. Las formas de presentación pueden ser leves, como hematomas, o más severos, como disecciones arteriales, aneurismas y roturas espontáneas. La forma más grave se manifiesta en el tipo vascular6, donde sus complicaciones son principalmente arteriales de vasos abdominales de tamaño mediano (renal, ilíaca, femoral, mesentérica y hepática)7 y la aorta abdominal, aunque también se han descrito en el tipo clásico. Los métodos diagnósticos invasivos deben evitarse ya que podrían causar graves complicaciones debido a la fragilidad de los vasos4. Se recomienda utilizar modalidades no invasivas como la ecografía, la angio-TC o la MRI. A diferencia de nuestra actuación, debe evitarse la angiografía, que se asocia con una tasa de complicaciones de entre el 17-67% y de una mortalidad del 6-19%8.
En el tratamiento quirúrgico de los trastornos vasculares en pacientes con EDS, actualmente están reconocidos tanto los procedimientos abiertos como los endovasculares con buenos resultados1, pero pocos pacientes se han sometido a tratamientos endovasculares de urgencia2. En caso de hemorragia se recomienda ser lo más conservador posible, mediante la administración de desmopresina, compresión y el uso de medidas hemostáticas locales y pegamentos9. El siguiente paso consiste en la utilización de modalidades endovasculares como la embolización o la reparación mediante stents o prótesis. Como ocurrió en nuestro caso, cuando fallan las medidas menos invasivas, se debe recurrir a la cirugía4.
Hasta el momento, los 5 casos descritos de rotura espontánea de la arteria esplénica secundaria a un aneurisma están asociados al EDS tipo vascular, con una mortalidad del 25%10, siendo el caso que presentamos el primero de rotura de arteria esplénica asociado al EDS tipo clásico de la literatura.
Con este caso intentamos destacar la importancia de una valoración global del paciente, donde tanto la historia clínica como la exploración física, nos permitan relacionar los hallazgos intraoperatorios con el diagnóstico final.

