




La granulomatosis de Wegener (GW), descrita por Klinger en 1931 como una variante de la poliarteritis nudosa y posteriormente descrita por Wegener en 1936, es una entidad sistémica infrecuente de causa desconocida caracterizada por la presencia de inflamación granulomatosa en el tracto respiratorio superior e inferior, vasculitis necrosante generalizada de los vasos de pequeño y mediano tamaño y glomerulonefritis. Se trata de una enfermedad compleja y de difícil diagnóstico en ausencia de la tríada clínica clásica1. La afectación de la mama es extremadamente inusual y en la mayoría de pacientes se produce después de otras manifestaciones sistémicas. Excepcionalmente puede ser el primer signo clínico y puede aparecer de manera aislada varios meses antes que el resto de las manifestaciones2. Presentamos un caso de GW con afectación mamaria sin signos iniciales de enfermedad sistémica.
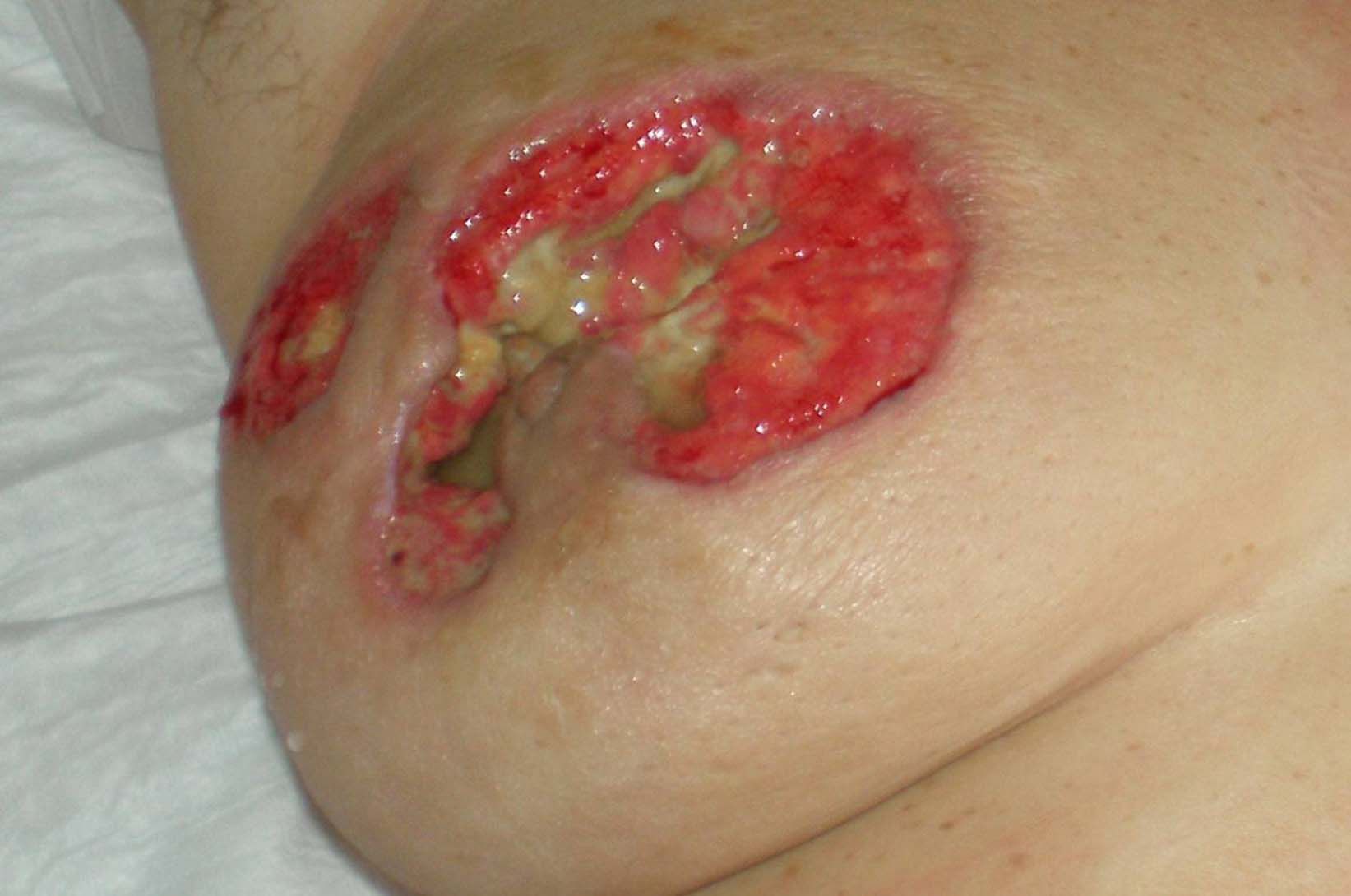
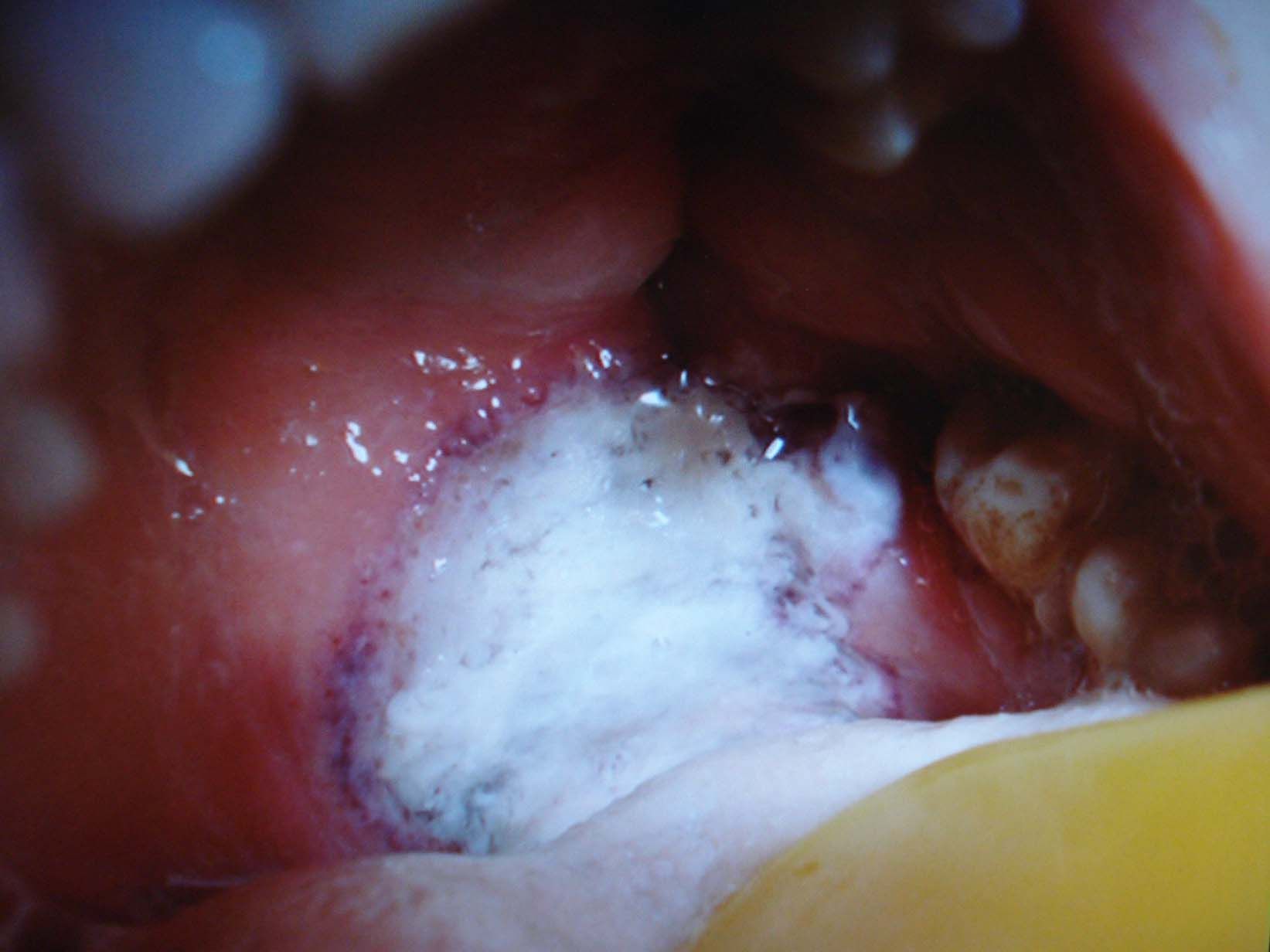
Una mujer de 53 años de edad, sin antecedentes de interés, excepto madre fallecida por una neoplasia mamaria, consultó por una historia de lesiones cutáneas de 10 meses de evolución. A la exploración física se observaba una úlcera cutánea en la mama derecha, dolorosa, de grandes dimensiones con una base purulenta y unos márgenes inflamatorios en sacabocados (fig. 1). Inicialmente, se trataba de pequeños nódulos subcutáneos que posteriormente se ulceraban y que se diagnosticaron clínica y ecográficamente como pequeños abscesos retroareolares en el contexto de una mastitis. La paciente recibió 2 ciclos de tratamiento antibiótico oral y 2 drenajes quirúrgicos durante esos meses. Los cultivos microbiológicos fueron negativos y el estudio histológico de las 3 biopsias realizadas mostró una dermatitis granulomatosa nodular inespecífica, con amplias áreas de necrosis y ausencia de células neoplásicas. La mastitis presentó una escasa respuesta al tratamiento antibiótico, y ante la sospecha de una neoplasia a pesar del resultado de las biopsias, finalmente se decidió realizar una mastectomía simple. La anatomía patológica mostró un parénquima mamario con un infiltrado inflamatorio crónico, con reacción gigantocelular a cuerpo extraño y tejido de granulación abscesificado. Dos semanas después, la paciente acudió a urgencias por un cuadro de fiebre elevada con poliartralgias, una intensa odinofagia debida a una ulceración necrosante en la orofaringe (fig. 2) y la aparición en ambos miembros inferiores de lesiones cutáneas de púrpura palpable. Los hallazgos histopatológicos fueron compatibles con una vasculitis leucocitoclástica de pequeño vaso. En el examen de laboratorio destacaba una elevación de la velocidad de sedimentación globular de 104mm/h, una proteína C reactiva de 21,92mg/dl, una hemoglobina de 10,4g/dl y una positividad para los anticuerpos anticitoplasma del neutrófilo (C-ANCA) frente a la proteinasa 3 de 305U/ml. Se detectó una microhematuria (40–60hematíes/μl), una proteinuria (0,3g/24h) que no presentaba previamente y un nódulo cavitado de 7,5mm en el lóbulo pulmonar superior izquierdo. Al establecerse el diagnóstico definitivo de GW, la paciente inició tratamiento con prednisona (60mg/día) y ciclofosfamida (100mg/día) durante los siguientes 4 meses con una completa resolución del cuadro clínico y una negativización de los auto-C-ANCA.


La GW afecta generalmente de forma agresiva a pacientes en su cuarta o quinta década de vida con una incidencia algo superior en varones3. Cualquier órgano puede afectarse y las lesiones cutáneas pueden ser el signo inicial de la enfermedad en aproximadamente un 10% de los casos. Su presencia puede indicar la progresión a una forma sistémica activa4, lo que lo convierte en un importante marcador pronóstico5. Aunque ninguna lesión cutánea de forma aislada es patognomónica, ciertos hallazgos son muy indicativos, como la púrpura palpable y las úlceras necrosantes6. Suelen aparecer en pacientes jóvenes que presentan un curso clínico más agresivo, con una afectación articular y renal hasta en el 80% de los casos7. La positividad de los C-ANCA ocurre en un 90% de las formas generalizadas activas y su determinación seriada puede ayudar a valorar la actividad de la enfermedad en un contexto clínico, analítico y radiológico determinado4. En aquellos pacientes con un diagnóstico de sospecha o definitivo de una vasculitis sistémica que asocian afectación mamaria se recomienda la realización de una biopsia de confirmación debido a la elevada prevalencia del cáncer de mama en la población femenina8.
Respecto al tratamiento, el combinado con corticoides y ciclofosfamida es el régimen más utilizado y provoca una remisión completa en el 75% de los enfermos9. Los procedimientos quirúrgicos están contraindicados dado que favorecen la aparición de nuevas úlceras cutáneas o el empeoramiento de las úlceras preexistentes3.
En conclusión, la afectación aislada de la mama en la GW sin otras manifestaciones sistémicas asociadas es extremadamente inusual. Puede iniciar como nódulos, masas o ulceraciones y puede generar un retraso en el diagnóstico, la realización de procedimientos quirúrgicos intensivos innecesarios y una progresión de la enfermedad con afectación de otros órganos. Tampoco es frecuente la afectación mamaria en el contexto de una GW, existen aproximadamente unos 20 casos publicados en la literatura médica en estas 4 últimas décadas10. La GW debería incluirse en el diagnóstico diferencial de las úlceras necrosantes de la mama con mala evolución clínica y hallazgos histológicos inespecíficos, de la mastitis granulomatosa y de otras neoplasias malignas de la mama.

