





La hipercolesterolemia familiar (HF) es una enfermedad autosómica dominante, causada principalmente por mutaciones en la región codificante del gen del receptor de las LDL (LDLR). Sin embargo, varias mutaciones situadas en el promotor de LDLR se han asociado con la HF. La búsqueda de mutaciones en sujetos clínicamente diagnosticados como HF reveló la presencia en la zona promotora de LDLR de 4 mutaciones nuevas en heterocigosidad.
ObjetivoEstudiar la funcionalidad de las 4 mutaciones nuevas en el promotor del LDLR (c.-36T>G, c.-136C>G, c.-140C>G y c.-208A>T) encontradas en España mediante el uso de la plataforma LIPOchip® en pacientes con sospecha clínica de HF.
MétodosSe realizó el análisis funcional de las mutaciones mediante ensayos de retardo de la movilidad electroforética (EMSA) y transfección de promotores mutados con el gen reportero de la luciferasa en cultivos celulares de HepG2.
ResultadosLas mutaciones c.-136G y c.-140G localizadas en el elemento regulador R3 mostraron un cambio significativo en el patrón de afinidad por las proteínas nucleares en los EMSA. Además, estas mutaciones produjeron una reducción de la actividad del promotor LDLR del 88-93%. Las mutaciones c.-36G y c.-208T no provocaron cambios significativos ni en los experimentos EMSA ni con genes reporteros.
ConclusionesLas mutaciones localizadas en el elemento R3 se asocian con HF, mientras que las que se encuentran fuera de los elementos reguladores del promotor de LDLR no son causa directa de hipercolesterolemia. Nuestros resultados revelan la importancia de realizar análisis de funcionalidad de las variantes encontradas en la región promotora de LDLR con objeto de conocer su implicación con el fenotipo HF.
Familial hypercholesterolemia (FH) is an autosomal dominant disorder mainly caused by mutations in the coding region of the LDLR gene. However, a variety of mutations within the LDLR promoter have been associated with FH. Genetic screening in persons clinically diagnosed with HF revealed the presence of four new heterozygous mutations within the promoter region.
ObjectiveTo study the functionality of the four new LDLR promoter mutations (c.-36T>G, c.-136C>G, c.-140C>G and c.-208A>T) found in Spain, using the LIPOchip® platform in patients with clinically suspected FH.
MethodsThe functional analysis of mutations was carried out by using electrophoretic mobility shift assays (EMSA) and luciferase reporter gene expression in HepG2 transfected cells with the mutated promoters.
ResultsTwo mutations, c.-136G and c.-140, located within the R3 regulatory element, showed a significant change in the pattern of nuclear binding protein affinity. Moreover, these mutations reduced the residual activity of the LDLR promoter by 88-93%. However, mutations c.-36G and c.-208T, located outside the response elements, produced no significant changes in EMSA experiments or reporter genes.
ConclusionsMutations within the R3 element are associated with FH, while those located outside the regulatory elements of the LDLR promoter are not a direct cause of FH. Our results reveal the importance of functional analysis of the new variants in the LDLR promoter region to identify their role in the FH phenotype.
La hipercolesterolemia familiar (HF) es una enfermedad autosómica dominante caracterizada por concentraciones muy elevadas de colesterol LDL (cLDL), xantomas tendinosos (XT) y riesgo elevado de padecer enfermedad coronaria prematura1. La HF fue descrita por primera vez por Müller en 19382, pero no es hasta mediados de los años setenta cuando Brown y Goldstein encontraron en el receptor de las LDL (rLDL) la causa de la enfermedad, y actualmente se conocen más de 1.000 mutaciones en el gen que codifica para el rLDL (LDLR) asociadas con HF (http://www.ucl.ac.uk/fh, http://www.umd.necker.fr)3,4. Posteriormente se han descrito mutaciones en el gen que codifica para la apolipoproteína B (APOB), ligando natural del rLDL5, y en PCSK9, que producen un fenotipo similar6. La mayoría de las mutaciones descritas en las bases de datos se concentran en la región codificante del gen LDLR; sin embargo, últimamente se han identificado varias mutaciones en los elementos reguladores del promotor de LDLR y en sitios de ayuste7.
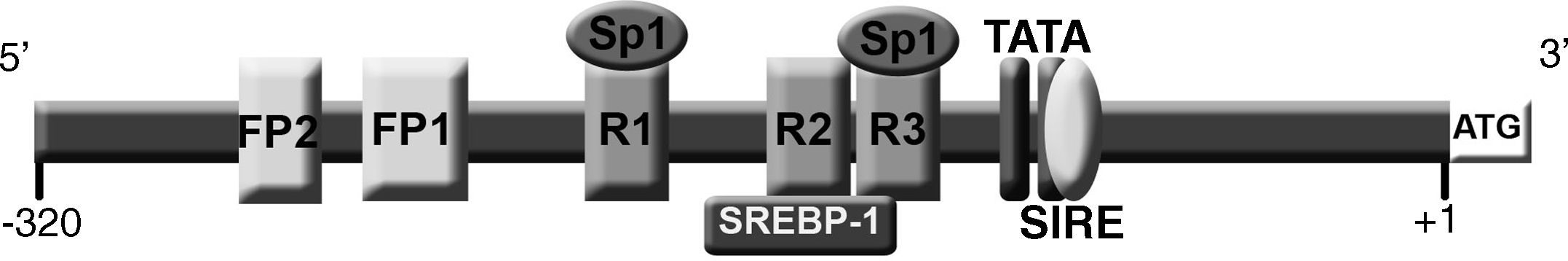
La síntesis proteica de rLDL está regulada de manera muy sofisticada por un mecanismo de retroalimentación que controla la transcripción del gen LDLR en respuesta a variaciones intracelulares de la concentración de esteroles y la demanda celular de colesterol8,9. Los elementos esenciales para la regulación transcripcional del LDLR se localizan a lo largo de 280 pb en el promotor, partiendo del sitio de inicio de transcripción. Esta región contiene todos los elementos para la expresión basal y la regulación por esteroles, incluyendo 3 repeticiones directas imperfectas de 16 pb cada una (R1, R2 y R3). Las regiones R1 y R3 contienen sitios de unión para el factor de transcripción Sp18 y contribuyen a la expresión basal que requiere la contribución de R2 para la sobreexpresión del gen10. La repetición 2 contiene un elemento de regulación por esteroles (SRE) que incrementa la transcripción cuando la concentración intracelular de esteroles es baja, al ser reconocido por el factor de transcripción SREBP-111. Entre las posiciones -110 a -94 de la región 5’UTR, y solapándose con las secuencias de las caja TATA-like, se reconoció un elemento cis independiente de la regulación por esteroles, denominado SIRE12. Además de estos elementos, se han descrito dos más, FP1 y FP2, localizados entre las posiciones -281 y -269, que contienen elementos esenciales para la inducción máxima de la transcripción (fig. 1)13. Recientemente se ha identificado un nuevo elemento rico en CT que se encuentra en la secuencia R3 e interactúa con el transactivador hnRNP K, una ribonucleoproteína nuclear que reconoce el elemento en la cadena sencilla y activa la transcripción del gen LDLR14.
. Las dos cajas TATA se sitúan entre las posiciones -101 y -116. Las repeticiones 1 y 3 (R1 y R3) localizadas entre las posiciones -188 a -196 y -130 a -145, respectivamente, son elementos de unión a Sp1 y contribuyen a la expresión basal del gen, y colaboran en la regulación mediada por esteroles. La repetición 2 (R2) se localiza entre las posiciones -146 a -161 y contiene el sitio de unión del factor de transcripción SREBP-1 regulado por la concentración de esteroles. Entre las posiciones -218 a -238 y -268 a -280 se encuentran los elementos de regulación FP1 y FP2.")
Representación gráfica de la región promotora y 5’UTR del genLDLR. El nucleótido A del ATG es considerado como la posición +1. Entre las posiciones -94 y -110 se encuentra el elemento de regulación independiente de los esteroles (SIRE). Las dos cajas TATA se sitúan entre las posiciones -101 y -116. Las repeticiones 1 y 3 (R1 y R3) localizadas entre las posiciones -188 a -196 y -130 a -145, respectivamente, son elementos de unión a Sp1 y contribuyen a la expresión basal del gen, y colaboran en la regulación mediada por esteroles. La repetición 2 (R2) se localiza entre las posiciones -146 a -161 y contiene el sitio de unión del factor de transcripción SREBP-1 regulado por la concentración de esteroles. Entre las posiciones -218 a -238 y -268 a -280 se encuentran los elementos de regulación FP1 y FP2.
Actualmente hay registradas varias mutaciones que se localizan en la región promotora del gen LDLR y que afectan a la función del rLDL. Se han descrito mutaciones en R2 (c.-153C>T)15 en el sitio de reconocimiento del factor de transcripción Sp1 en la secuencia R3 (c.-142C>T, c.-138T>C, c.-138delT, c.-137C>T, c.-136C>G)16–20 y en la región próxima a la caja TATA (c.-120C>T)15 que se asocian con el fenotipo HF. Incluso se ha demostrado que las mutaciones descritas en los elementos reguladores FP1 y FP2 provocan una alteración en la actividad transcripcional de LDLR21.
El objetivo de este trabajo fue realizar el análisis funcional de 4 mutaciones nuevas en el promotor del LDLR encontradas en España mediante el uso de la plataforma LIPOchip® en pacientes con sospecha clínica de HF22.
Material y métodosDetección de mutacionesLos sujetos considerados como probandos para este estudio fueron diagnosticados clínicamente según los criterios MedPed, considerándose aquellos con diagnóstico definitivo de HF (individuos cuya puntuación fuera mayor de 8) o probable (puntuación entre 6-8)23. Partiendo de muestras de sangre periférica se extrajo el ADN mediante el producto QiAmp Blood DNA Mini Kit (Qiagen, Alemania), según las instrucciones del fabricante y posteriormente analizado con la plataforma LIPOchip® (Progenika Biopharma, Bilbao, España)22. Para aquellos sujetos en los que no se identificó la mutación mediante el método de microarray, se secuenciaron tanto el promotor, como los exones y las regiones de unión exón-intrón del LDLR.
Ensayos de retardo de la movilidad electroforética (EMSA)Para la realización de los ensayos de retardo de la movilidad electroforética se diseñaron los siguientes oligonucleótidos de doble cadena marcados durante la síntesis con IRDye700 (Li-Cor Biosciences, Lincoln, NE): GCAGGTCGTGA(T/G)CCGGGTCGG (para c.-36T>G), CAAACTCCTC(C/G)CCCTGCTAG (c.-136C>G), CACTGCAAACT(C/G)CTCCCCCTGC (c.-140C>G) y CCGATGTCAC(A/T)TCGGCCGTT (c.-208A>T). La obtención de los extractos nucleares y el procedimiento de la electroforesis se han descrito en trabajos anteriores24. El análisis y la cuantificación de los EMSA se realizaron con el Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln, Nebraska).
Construcciones de plásmidos con el promotor del gen LDLR y el gen reportero luciferasaLos constructos fueron diseñados en 2 tipos diferentes de vectores GENEART (Geneart AG, Alemania) resistentes a la ampicilina. Para el constructo que contiene la secuencia salvaje y los constructos portadores de las mutaciones c.-120T, c.-135G, c.-136G y c.-208T, partiendo de los vectores GENEART, se amplificó un fragmento de 271 pb del promotor de LDLR, desde la posición -319 a -64, usando los cebadores siguientes: 5’-CTGAGCTCCAGCTCTTCACCGGAGACC-3’ y 5’-CTGCTAGCCCTGCTGTGTCCTAGCTGGAAA-3’. Para las mutaciones c.-36G y c.-140G, así como para la secuencia salvaje, se amplificó un fragmento de 325bp del promotor, desde la posición -319 a -1 usando el cebador directo anteriormente descrito y el reverso: 5’-GCTAGCGCTCGCAGCCTCTG-3’. Estos oligonucleótidos introducen la secuencia de reconocimiento para las enzimas de restricción SacI y NheI (subrayado) en los extremos 5’ y 3’, respectivamente, del producto resultante. Se purificó el producto de PCR mediante el QIAprep Spin Miniprep Kit (Qiagen Inc, Chatworth, California). El plásmido purificado fue digerido con las enzimas de restricción anteriormente nombradas y subclonado en el vector pGL3-Basic (Promega Madison, Wisconsin) que contiene el gen reportero de la luciferasa y carece de promotor24. Se verificó la presencia de las variantes adecuadas y ausencia de otras mutaciones introducidas por PCR mediante secuenciación del plásmido resultante.
Cultivos celularesSe cultivaron células HepG2 en medio DMEM (Dulbecco's Modified Eagle's Medium) suplementado con 100 U/ml de penicilina, 100μg/ml de estreptomicina y 10% de suero fetal bovino (SFB). Para los experimentos de transfección, se sembraron células HepG2 con densidad de 1×106 células/pocillo de una placa de 12 pocillos y se dejaron crecer hasta alcanzar la confluencia en 1ml de DMEM con y sin SFB, en dependencia del experimento. Dichas células fueron transfectadas mediante Lipofectamine 200™ (Invitrogen, Carlsbad, California) con 1,6μg del constructo reportero de pGL3-BASIC y 0,05μg de pRL-TK para normalizar los experimentos, según las instrucciones del fabricante. Tras 36h, se lisaron las células y se realizaron los experimentos para medir la actividad de luciferasa usando el producto Dual-Luciferase® Reporter Assay (Promega, Madison, Wisconsin). Los resultados obtenidos fueron integrados y analizados mediante el programa informático WinGlow v.124 (Berthold Technologies GmbH & Co. KG, Bad Wildbad, Alemania). Se normalizaron los datos respecto a la actividad específica de Renilla. Cada experimento fue realizado por triplicado.
ResultadosDetección de mutacionesTras el análisis llevado a cabo por la plataforma LIPOchip® (Progenika Biopharma, Bilbao, España) en individuos con un diagnóstico probable o definitivo de FH, según los criterios MedPed23, se identificaron 6 sujetos que carecían de mutaciones frecuentes detectadas por el chip, pero, tras la secuenciación de las regiones codificantes, las uniones exón-intrón, así como el promotor, presentaron mutaciones en heterocigosis en la región promotora. Dos de las variantes encontradas en esta población, c.-120C>T y c.-135C>G, ya habían sido descritas y analizadas funcionalmente por otros estudios15,18.
Considerando las 4 mutaciones nuevas, 2 de ellas se localizaban en la zona de reconocimiento para el factor de transcripción Sp1, dentro de R3: c.-136C>G y c.-140C>G8. Las restantes variantes no se encontraban dentro de los elementos reguladores.
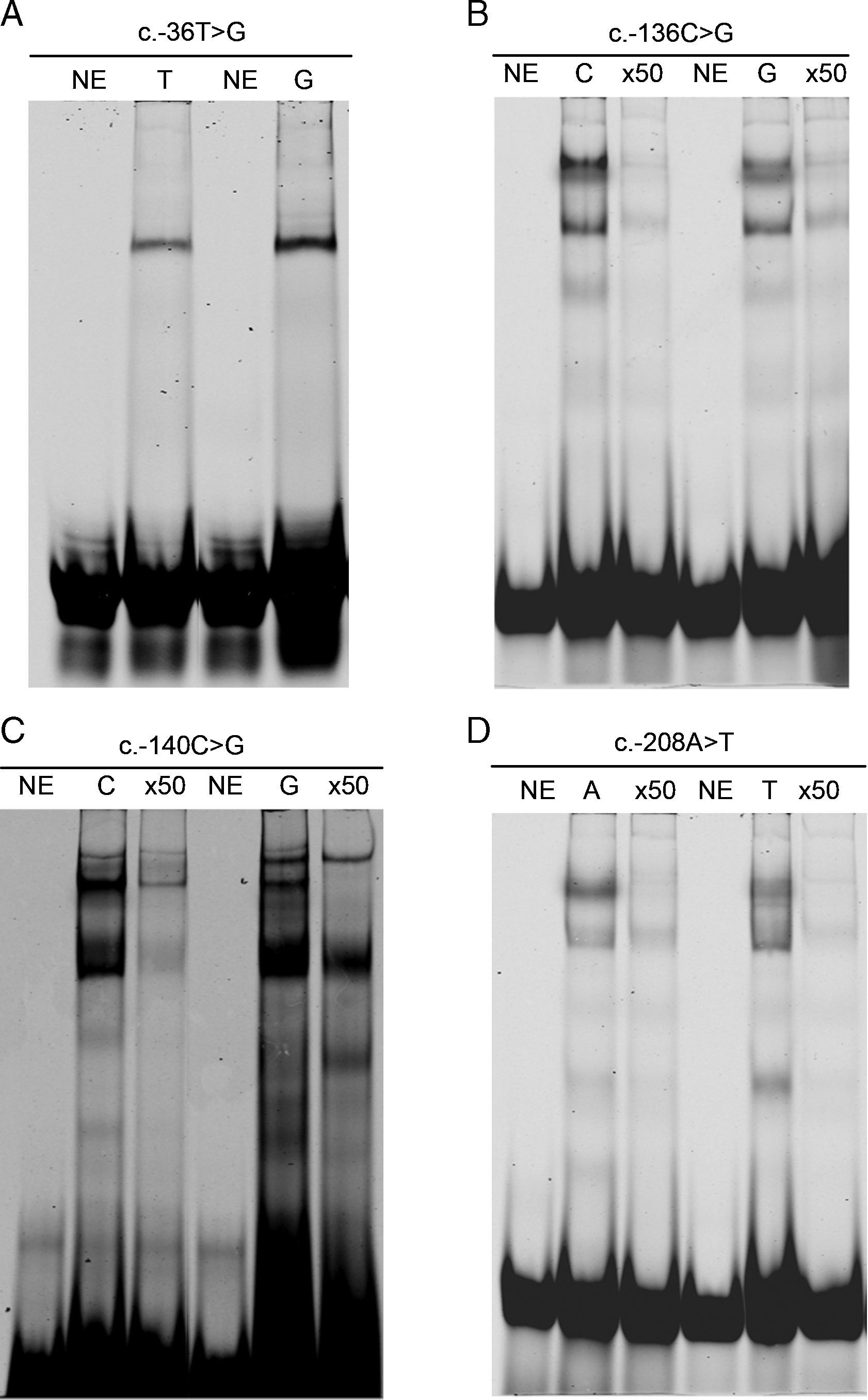
Estudio funcional de las mutacionesCon objeto de analizar la implicación de las nuevas mutaciones en la funcionalidad del promotor del gen LDLR, realizamos ensayos de retardo de la movilidad electroforética. Como se observa en la figura 2 (B y C), las mutaciones de R3, c.-136G y c.-140G, produjeron una reducción en la afinidad de los oligonucleótidos por las proteínas nucleares, así como una reducción del 88 y 93%, respectivamente, en la actividad luciferasa analizada mediante los constructos mutantes del promotor de LDLR en plásmidos pGL3 (tabla 1). Sin embargo en los EMSA y la actividad transcripcional de LDLR que se encontraban fuera de las regiones descritas como reguladoras correspondientes a las variantes c.-36G y c.-208T no mostraron cambios significativos (fig. 2 A y D).
 Efecto de las mutaciones c.- 36T>G y c.-208A>T sobre las propiedades de unión a proteínas de fragmentos de ADN. Se mezclaron 0,5μl de oligonucleótidos (10μM) marcados con un fluoróforo, con 20μg de extracto nuclear. La mutación analizada se indica en la parte superior de cada panel. Carriles: NE indica que el experimento se ha llevado a cabo en ausencia de extracto nuclear. T, G o A indican la identidad del alelo. x50 indica el exceso de oligonucléotido marcado que ha sido añadido a la mezcla antes de la reacción de unión. B y C) Efecto de las mutaciones c.-136C>G y c.-140C>G sobre las propiedades de unión a proteínas de fragmentos de ADN. La mutación analizada se indica en la parte superior de cada panel. Carriles: NE indica que el experimento se ha llevado a cabo en ausencia de extracto nuclear. C, G, T o A indican la identidad del alelo. x50 indica el exceso de oligonucléotido marcado que ha sido añadido a la mezcla antes de la reacción de unión.")
A y D) Efecto de las mutaciones c.- 36T>G y c.-208A>T sobre las propiedades de unión a proteínas de fragmentos de ADN. Se mezclaron 0,5μl de oligonucleótidos (10μM) marcados con un fluoróforo, con 20μg de extracto nuclear. La mutación analizada se indica en la parte superior de cada panel. Carriles: NE indica que el experimento se ha llevado a cabo en ausencia de extracto nuclear. T, G o A indican la identidad del alelo. x50 indica el exceso de oligonucléotido marcado que ha sido añadido a la mezcla antes de la reacción de unión. B y C) Efecto de las mutaciones c.-136C>G y c.-140C>G sobre las propiedades de unión a proteínas de fragmentos de ADN. La mutación analizada se indica en la parte superior de cada panel. Carriles: NE indica que el experimento se ha llevado a cabo en ausencia de extracto nuclear. C, G, T o A indican la identidad del alelo. x50 indica el exceso de oligonucléotido marcado que ha sido añadido a la mezcla antes de la reacción de unión.
Valores en porcentaje de la actividad luciferasa relativa de los promotores mutantes de LDLR
| Mutación | % Actividad luciferasa relativa mutante vs. salvaje | |
| p | ||
| c.-36T>G | 128±73,9 | 0,387 |
| c.-120C>Ta | 52±15.8 | 0,001* |
| c.-135C>Gb | 11±5,3 | <0,0001* |
| c.-136C>G | 12±10,9 | <0,0001* |
| c.-140C>G | 7±2,6 | <0,0001* |
| c.-208A>T | 84±40,2 | 0,116 |
Valores de la actividad luciferasa relativa expresados como media de los porcentajes obtenidos en tres experimentos individuales desarrollados por triplicado.
Las mutaciones c.-120C>T y c.-135C>G habían sido previamente analizadas por Francova et al15 y Hobbs et al18, respectivamente. Para comprobar la metodología de los ensayos de funcionalidad utilizados en este trabajo, se volvieron a analizar, y tal y como se esperaba mostraron una reducción de la actividad del promotor del 48% (p<0,001) y el 89% (p<0,0001), respectivamente (tabla 1).
DiscusiónEn este trabajo analizamos 4 mutaciones en el LDLR no descritas previamente, y dos de ellas se localizaron en la principal región de regulación8. La mayoría de las secuencias reguladoras del gen LDLR se encuentran en los 280 nucleótidos anteriores al codón de iniciación. En esta región se localizan dos secuencias ricas en AT, denominadas cajas TATA, y tres repeticiones imperfectas de 16 nucleótidos que son responsables de la actividad del promotor, así como un elemento de regulación independiente de esteroles (SIRE) que se encuentra entre las posiciones -94 y -110, incluye una de las cajas TATA y contiene los sitios de unión para C/EBP y CREB (fig. 1)8,12. La transcripción de LDLR es estimulada por los esteroles a través del SREBP y su sitio de unión SER-1 localizado en la repetición 29–11. Además, las repeticiones 1 y 3 son elementos de unión a Sp1 para contribuir a la expresión basal del gen, y cooperan con la repetición 2 en la regulación mediada por esteroles10. Recientemente se ha identificado una proteína transactivadora, hnRNP K, que reconoce el elemento rico en CT de la región R314.
Estudios de funcionalidad y co-segregación han demostrado que otras mutaciones que se producen en los nucleótidos conservados de R3 pueden considerarse causales del fenotipo HF. Es en esta región donde se han descrito la mayoría de las mutaciones identificadas en el promotor hasta ahora25. La mayoría de las mutaciones en esta zona producen amplias reducciones en la potencia del promotor, obteniéndose generalmente entre el 5 y el 15% de la actividad transcripcional del promotor tipo salvaje16–20,26,27. Los resultados obtenidos para las mutaciones identificadas en esta región a lo largo de este trabajo, c.-136C>G y c.-140C>G, correspondieron con lo esperado. Los experimentos EMSA realizados mostraron un patrón de afinidad por las proteínas nucleares diferente según se utilizase el alelo mutado o el salvaje (fig. 1). Igualmente, los experimentos de transfección de clones mutantes en cultivos celulares de HepG2 presentaron una actividad residual del promotor LDLR del 12 y del 7%, respectivamente (tabla 1). Estos valores fueron similares a los que observó el grupo de Hobbs et al18 con la mutación c.-135C>G, que nosotros utilizamos como control interno y con la que obtuvimos una actividad residual del 11%.
La mutación c.-120C>T también fue utilizada como control interno, con el fin de tener un valor orientativo de la afectación en la actividad transcripcional por parte de mutaciones no localizadas en regiones reguladoras principales. Nuestro grupo obtuvo una reducción del 48% en la actividad del promotor de LDLR, valores similares a los obtenidos por Francova et al15.
Los resultados obtenidos en los EMSA para el resto de variantes, c.-36T>G y c.-208A>T, indicaron que sólo producían cambios minoritarios en el patrón de bandas que determina la afinidad de una secuencia por las proteínas nucleares (datos no mostrados). El análisis de la actividad transcripcional del promotor LDLR mutado para con estas variantes no mostró diferencias significativas (tabla 1). Aunque estas mutaciones no se encuentran en las regiones más importantes de regulación, se ha demostrado que la variante c.-217C>T, localizada cerca del nucleótido c.-208, produce cambios en la actividad transcripcional del promotor LDLR10. Se han identificado muy pocas mutaciones en la región 5’UTR, una de las cuales, c.-22delC, creaba un nuevo codón de inicio de la traducción y era por tanto causante de la HF28. Sin embargo, los pequeños cambios que producen las mutaciones externas a las zonas de regulación sugieren que, en general, aunque podrían tener cierta influencia sobre los niveles del colesterol, su efecto no sería suficiente para ser causa del fenotipo HF de los pacientes.
En conclusión, hemos encontrado 4 mutaciones nuevas en el promotor del gen LDLR de varios pacientes afectos de HF. Los estudios de funcionalidad demuestran que sólo 2 de ellas, que se localizan en la repetición 3, producen efectos suficientemente drásticos para considerarlas responsables de la HF. Estos resultados muestran la necesidad de realizar análisis de funcionalidad en las nuevas variantes encontradas en el promotor del gen LDLR, con el fin de conocer su influencia en el fenotipo HF.
Contribución de los autoresIsabel de Castro-Orós ha participado en el diseño, la recogida de datos, el análisis y la interpretación de los mismos, así como en la realización del borrador del artículo y la aprobación final del mismo.
Sandra Pampín, Alfonso Bolado-Carrancio y Lourdes Palacios participaron en el diseño, la recogida, el análisis y la interpretación de los resultados, colaboraron en la realización del borrador del artículo y aprobaron el resultado final.
Aguirre de Cubas, Nuria Plana, Esperanza Martorell, José Puzo y Luis Masana han participado en el diseño, en la recogida y el análisis de datos, en la revisión crítica del contenido del artículo y en su aprobación final.
Marianne Stef, Fernando Civeira, José Carlos Rodríguez-Rey y Miguel Pocoví diseñaron el estudio, realizaron una revisión crítica del contenido del artículo y aprobaron el resultado final.
FinanciaciónEste estudio ha sido financiado con una beca de la Sociedad/Fundación Española de Arteriosclerosis para Investigación Básica (2005).
Conflicto de interesesLos autores declaran que no tienen conflictos de intereses.