






La hiperlipemia familiar combinada (HFC) es la hiperlipemia genética más frecuente en pacientes supervivientes a infarto de miocardio (IM) antes de los 60 años. Aunque la HFC fue descrita por Goldstein et al, en 19731, esta enfermedad presenta un complejo fenotipo que no está totalmente esclarecido. Los pacientes presentan elevaciones del colesterol total y/o triglicéridos séricos que pueden variar a lo largo del tiempo, dando fenotipos diferentes según la clasificación de Fredrickson2: IIa cuando presentan hipercolesterolemia, IV cuando presentan hipertrigliceridemia o IIb cuando la hiperlipemia es mixta. También se observa en estos pacientes una elevación de la apolipoproteína B100 (Apo B) y Apo CIII. Aparece con una prevalencia de aproximadamente del 1 al 2% en la población general3 y en pacientes con cardiopatía isquémica alrededor del 14% presentan esta enfermedad.
La HFC no tiene ninguna prueba diagnóstica específica, lo que dificulta su diagnóstico preciso. Bredie et al4 han propuesto una serie de criterios de inclusión y exclusión que son los más utilizados (tabla 1).

Diagnóstico
Tres criterios mayores de inclusión y exclusión; o un criterio mayor de inclusión y dos criterios de exclusión, y al menos un criterio de inclusión adicional.
Aunque el carácter hereditario de esta alteración es incuestionable, no se sabe claramente cuál es el defecto genético causante; los estudios en familias afectadas parecen poner de manifiesto una herencia autosómica dominante con alta penetrancia. Debido a esta transmisión se pensó en principio que se trataría de una alteración monogénica, pero la gran heterogeneidad, tanto genotípica como metabólica, que presentan los pacientes y sus familias hace que se piense más en una herencia poligénica en la que los desórdenes resultan de uno o más genes e interacciones genes-ambiente.
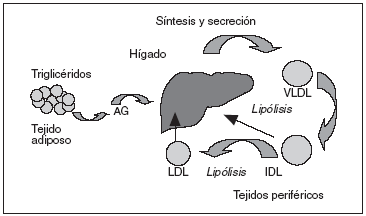
La patogenia de la enfermedad está relacionada con anormalidades en el metabolismo de las lipoproteínas ricas en triglicéridos5, incluida una hipersecreción hepática de lipoproteínas de muy baja densidad (VLDL) y Apo B, así como un retraso en el aclaramiento de las lipoproteínas ricas en triglicéridos (VLDL y quilomicrones remanentes). Incrementos hepáticos en la secreción de las VLDL contribuyen a una elevación de triglicéridos, Apo B y colesterol total en el plasma. Se ha sugerido recientemente que una hipersecreción VLDL1 y VLDL2 y un incremento del tiempo de residencia de las VLDL1 en la circulación contribuyen a la hipertrigliceridemia en la HFC (fig. 1). Anormalidades en el metabolismo lipídico e insulinorresistencia en el tejido adiposo y muscular son otras de las características de la HFC. Los altos valores de ácidos grasos libres (AGL) en la circulación conducen a una disminución en el estímulo de insulina y a un incremento de las lipoproteínas hepáticas, ambos típicos de la HFC. Por tanto, hígado, tejido adiposo y músculo son tejidos diana para el estudio de la expresión génica.
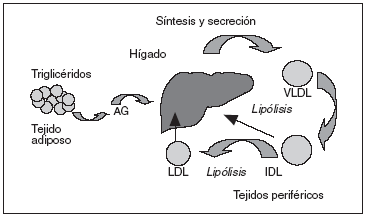
Figura 1. Modelo para la patogenia de la hiperlipemia familiar combinada. AG: ácidos grasos; IDL: lipoproteínas de densidad intermedia; LDL: lipoproteínas de baja densidad; VLDL: lipoproteínas de muy baja densidad.
Se puede utilizar un cribado del genoma6-8 para la identificación de regiones cromosómicas que contienen genes que contribuyen a la expresión del complejo fenotipo de la HFC, y estudios de genes candidatos, basándose en su función biológica, que puedan contribuir a aclarar la base genética de la HFC, aunque estos genes posiblemente no sean la primera causa de la enfermedad (tabla 2).

Lipoproteinlipasa
La lipoproteinlipasa (LPL) es la enzima clave en el catabolismo de las partículas ricas en triglicéridos (Qm y VLDL)9; hidroliza los triglicéridos y favorece el intercambio de apolipoproteínas y lípidos con las lipoproteínas de alta densidad (HDL), y transforma los Qm, y sobre todo las VLDL, en partículas cada vez más pequeñas y cargadas de colesterol. La alteración en este catabolismo da lugar a la aparición de partículas VLDL más pequeñas y con concentraciones más elevadas de Apo B, que condicionan también la aparición de un mayor número de partículas LDL pequeñas y densas (fenotipo B de las LDL). Se ha comprobado que estas partículas son más aterogénicas y con menor afinidad por el receptor de las LDL. La LPL se sintetiza preferentemente en las células del tejido muscular y adiposo. Después de su síntesis la enzima sufre una serie de modificaciones que van a controlar posteriormente su actividad. Así, si no existe una glucosilación, la enzima es inactiva. Tras las modificaciones se transporta a la parte externa de la membrana de las células endoteliales que rodean la luz de los capilares sanguíneos, donde se encuentran normalmente adheridas a proteoglicanos heparansulfato, y desde esta posición ejercen su función. También existe LPL libre en el plasma, pero ésta es inactiva. Los principales sustratos de la LPL son los Qm y VLDL, que además proveen a la LPL de un cofactor importante para su acción enzimática: la Apo CII.
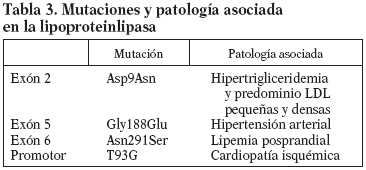
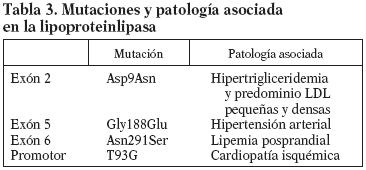
El gen de la LPL se localiza en el brazo corto del cromosoma 8p22 y está formado por 10 exones y 9 intrones con un tamaño de 30 Kb. El cADN codifica una proteína de 448 aminoácidos con un peso molecular (PM) de 50.394 D y su centro catalítico lo forman 3 aminoácidos: Ser132, Asp156 y His24110. Se han descrito unas 80 mutaciones del gen de la LPL en humanos11, la mayoría sin significado; las más frecuentes se encuentran en los exones 5 y 6. Muchas de estas mutaciones son poco frecuentes, aunque algunas están asociadas a la HFC y a enfermedades cardiovasculares prematuras (tabla 3). La mutación Asn291Ser situada en el exón 6 es una de las más frecuentes y ha aparecido asociada a aterosclerosis coronaria prematura y HFC. Algunos datos sugieren que esta mutación afectaría especialmente a la lipemia posprandial, empeorando el fenotipo de la HFC. La frecuencia de esta mutación en la población caucásica oscila en un 2-5%12. Otra mutación encontrada con relativa frecuencia en el gen de la LPL es la Asp9Asn, situada en el exón 2, que parece estar relacionada también con un aumento del riesgo de aterosclerosis coronaria, hipertrigliceridemia y aumento de LDL pequeñas y densas. Su frecuencia se ha estimado en un 4-5%13. Una tercera mutación encontrada ha sido Gly188Glu, situada en el exón 5 y que está relacionada con la hipertensión arterial14. Una cuarta mutación es la variante T93G, variante del promotor, normalmente en desequilibrio de ligamiento con la mutación Asp9Asn; esta doble heterocigosidad está asociada a elevadas concentraciones plasmáticas de triglicéridos y a un incremento de cardiopatía isquémica15. La presencia de estas mutaciones se puede analizar mediante amplificación por reacción en cadena de la polimerasa (PCR) de las regiones del gen en las cuales se encuentran localizadas16. Posteriormente, se realiza una digestión con enzimas de restricción Taq I, AvaII, Rsa. Los fragmentos se separan por electroforesis en gel de poliacrilamida (PPA) del 8%, lo que nos permite identificar a los portadores de estas mutaciones.
Apolipoproteína E
La apolipoproteína (Apo E) es una glicoproteína sintetizada fundamentalmente en el hígado, y en el plasma se encuentra en los Qm, VLDL y HDL. Es una proteína compuesta por 299 aminoácidos y codificada por un gen situado en el cromosoma 19q13 con una longitud de 3,7 Kb y 4 exones17. La naturaleza polimórfica del gen de la Apo E se describió tras los estudios de Uterman por isoelectroenfoque18. Es un gen con 3 alelos codominantes (E2, E3 y E4) que dan lugar a 6 posibles genotipos: E2/E2, E3/E3, E4/E4, E2/E3, E2/E4 y E3/E4. La frecuencia de los alelos E4, E3 y E2 en la población caucasiana obtenidos por electroforesis en gel de acrilamida es del 14, 80 y 6%, respectivamente19. Estas isoformas se diferencian en la sustitución de uno de los aminoácidos en la posición 112-15820. La E2 tiene cisteína en ambos, la E4 arginina en ambos y la E3 cisteína en el 158 y arginina en el 11221, lo cual modificará el reconocimiento de sus receptores específicos, especialmente en el caso de la Apo E2 (tabla 3).
El papel principal de la Apo E en el metabolismo lipoproteico es el aclaramiento por parte del hígado de las lipoproteínas ricas en triglicéridos (VLDL y Qm) y de sus remanentes, ya que actúan como ligando para el receptor hepático de las LDL y para el receptor de los remanentes (LRP)22. Pero, además, también regula la producción hepática de las partículas VLDL y la lipólisis de éstas por parte de la LPL. Los portadores de la isoforma E2 tienen menos colesterol intracelular debido a la escasa afinidad que tienen por el receptor de las LDL, lo que disminuye la captación hepática de Qm y VLDL23. Esto provoca un aumento en el número de receptores de LDL en el hepatocito. Además parece ser que la Apo E2 disminuye la conversión de VLDL en LDL. Por su parte, la isoforma E4 tiene una mayor afinidad por las VLDL y los Qm, lo que implica un aumento de colesterol intracelular que da lugar a una disminución de la expresión de los receptores de la LDL24. En el estudio Framingham se ha encontrado que en varones no sólo el alelo E4 se asocia con mayor riesgo de enfermedad cardiovascular, sino que también los portadores del alelo E2 tenían un significativo mayor riesgo25. El impacto de las diferentes isoformas de la Apo E, especialmente E2 y E4, en el metabolismo lipídico parece intervenir en la expresión fenotípica de la HFC.
El procedimiento para obtener el genotipo de la Apo E consiste en amplificar por PCR la zona del exón 426 y la posterior digestión con la enzima de restricción HhaI27, que nos permitirá distinguir si los nucleótidos incluidos en los codones 112 y 158 codifican Arg o Cys.
Cluster AI-CIII-AIV
El cluster AI-CIII-AIV, también codificado por genes situados en el brazo corto del cromosoma 11q23, contribuye al fenotipo de la HFC, aunque esta contribución es genéticamente compleja. La agrupación génica está constituida por unos 45 Kb. Los genes AI y AIV se transcriben en la misma dirección, mientras que el gen para la Apo CIII se transcribe en dirección contraria.
La Apo AI es el componente proteico mayoritario de las HDL29 y desempeña un papel fundamental en el transporte reverso de colesterol. La Apo CIII30 regula la lipólisis de las proteínas ricas en triglicéridos mediante su capacidad de inhibir la LPL. Así, concentraciones elevadas de esta proteína se asocian con un retraso en la eliminación de Qm posprandial. Por su parte, la Apo AIV tiene un papel menos definido, aunque se especula que puede ser un ligando para el receptor de HDL y que modula la actividad de la LCAT y LPL31. El cluster tiene un papel modulador en la HFC mediante el control de las concentraciones plasmáticas de las proteínas que codifica, especialmente de la Apo CIII, que suele estar elevada en estos pacientes. En la región intergénica situada entre CIII y AIV, que contiene los promotores de ambos genes, existe un gran número de dianas reguladoras que afectan a la totalidad del agregado genético32. Parece ser el cluster el causante de aproximadamente el 25% de la variabilidad de la concentración de HDL.
Se han estudiado principalmente 3 polimorfismos en el cluster: XmnI (C2500 T), MspI (G78A), ambos situados en la región 5' del gen de la apo AI y SstI (G3175 C) situado en la región no codificante del exón 4 del gen de la Apo CIII33. El polimorfismo XmnI contribuye a la variación plasmática de colesterol total y triglicéridos34. El polimorfismo MsPI está localizado en la región promotora, parece estar asociado con elevaciones en el plasma de la Apo AI y HDL y también contribuye a la variación del contenido de Apo B en el plasma. El polimorfismo SstI se ha asociado con hipertrigliceridemia y elevadas concentraciones de Apo CIII. La específica combinación de los haplotipos, 1-1-2/2-2-1 (1 = ausencia de sitio de restricción; 2 = presencia de sitio de restricción), representa la primera identificación de una región génica con alta susceptibilidad de HFC, agravándose la hipercolesterolemia y la hipertrigliceridemia, así como valores elevados de Apo CIII y Apo B35.
Para determinar la existencia de estos polimorfismos utilizamos la técnica de amplificación por PCR y la posterior digestión con las enzimas de restricción XmnI, MspI y SstI19.
Lipasa hepática
El gen de la lipasa hepática (LH) se localiza en el brazo largo del cromosoma 15q2136 y está constituido por 30 kb con 9 exones y 8 intrones. Este gen sintetiza una proteína de 477 aminoácidos con un peso molecular de 53 kD37. La LPL y la lipasa pancreática (LP) constituyen una familia de lipasas con un gen ancestral común.
La LH es una enzima que se sintetiza en el retículo endoplásmico de las células del hígado y reside en la superficie de éstas. Tiene 2 actividades: fosfolipasa y triglicérido lipasa, e interviene en el modelado de partículas IDL para su transformación en LDL, eliminando fosfolípidos y triglicéridos en los pasos finales38. También elimina fosfolípidos y triglicéridos de las HDL, provocando un aclaramiento de HDL del plasma, así como la conversión de HDL2 a HDL3, y su deficiencia da lugar a un patrón lipídico en el que aumentan el colesterol y los triglicéridos; el aumento de colesterol se debe fundamentalmente a la elevación de HDL. Variaciones en la actividad de la LH pueden predisponer a enfermedad arterial coronaria por su efecto sobre el tamaño y la densidad de las partículas LDL y HDL39. La deficiencia de LH se transmite de forma recesiva y es muy poco frecuente. Se han descrito algunas mutaciones en casos raros de déficit familiar de LH, que se asocian con pérdida de actividad de la enzima o con una alteración en su procesamiento celular. Se ha descrito una familia canadiense donde los pacientes son heterocigotos para 2 mutaciones: Ser627Phe y Thr383Met40. La primera mutación produce inactivación de la lipasa y la segunda una alteración en su secreción.
De los 4 polimorfismos encontrados en la zona del promotor y que se encuentran en completo desequilibrio de ligamiento, el más estudiado en relación con la HFC es el NlaIII (C514T)41, que parece explicar un 38% de la variabilidad en la actividad de la LH42. Los heterocigotos para esta mutación se relacionan con un aumento moderado de los valores de HDL en varones, pero no en mujeres. Los homocigotos se asocian a un marcado incremento de la concentración de HDL y Apo AI43.
En resumen, aunque no hay ninguna prueba diagnóstica específica para la hiperlipemia familiar combinada, el estudio de posibles genes candidatos, como los expuestos anteriormente, sería útil para comprender mejor la patogenia de la enfermedad y su expresión fenotípica, consecuencia de posibles interacciones genes-ambiente, así como para contribuir a un adecuado tratamiento.