





La mayoría de las hipertrigliceridemias (HTG) primarias graves se diagnostican en la edad adulta, y sus bases moleculares no se han dilucidado completamente.
Varios son los genes relacionados con este tipo de HTG, entre ellos el gen LMF1, que codifica la proteína Lmf1, la cual participa en la función de la lipoproteína lipasa (LpL). Teniendo en cuenta estos hechos, nuestro objetivo es identificar las variantes génicas comunes y no comunes en el gen LMF1 en sujetos con HTG primaria.
Hemos secuenciado el promotor, los exones y las regiones exón-intrón del gen LMF1 en 112 pacientes con HTG primaria grave, definida por triglicéridos por encima de 500mg/dl, excluyendo causas secundarias. Cinco pacientes (4,46%) fueron portadores de 4 variantes raras en LMF1 asociadas previamente a HTG. Además, se identificaron 2 variantes comunes con una frecuencia alélica diferente de la que se observa en población general: c.194-28 T>G y c.729+18C>G.
Se llevó a cabo un análisis bioinformático de las variantes encontradas, identificando las variantes p.Arg364Gln, p.Arg451Trp, p.Pro562Arg y p.Leu85Leu como potencialmente dañinas.
Nuestros resultados sugieren que el gen LMF1 contribuye a la etiología de la HTG primaria grave en un porcentaje significativo de los pacientes, con una combinación de mutaciones de efecto entre moderado y agresivo y polimorfismos clásicamente asociados con esta dislipidemia.
The majority of severe primary hypertriglyceridemia (HTG) are diagnosed in adults, and their molecular bases have not yet been fully defined.
The promoter, coding regions and intron-exon boundaries of LMF1 were sequenced in 112 patients with severe primary hipertrigliceridemia (defined as TG above 500mg/dl). Five patients (4.46%) were carriers of four rare variants in the LMF1 gene associated with HTG, which participate in lipoprotein lipase (LpL) function. Also, we have identified two common variants, c.194-28 T>G and c.729+18C>G that were associated with HTG, with a different allelic frequency to that observed in the general population.
A bioinformatic analysis of all found variants was conducted, defining the following as potentially harmful: p.Arg364Gln, p.Arg451Trp, p.Pro562Arg and p.Leu85Leu.
Our results suggest that LMF1 mutations are involved in a substantial proportion of cases with severe HTG, putting together the moderate-aggressive effect of rare mutations with polymorphisms classically associated with this disease.
Las hipertrigliceridemias (HTG) son un grupo heterogéneo de enfermedades del metabolismo lipídico que se caracterizan por una elevada concentración, en ayunas, de quilomicrones, lipoproteínas de muy baja densidad (VLDL) o sus remanentes. Su importancia clínica viene dada por su elevada frecuencia, y por su asociación con el desarrollo de pancreatitis, enfermedad cardiovascular, diabetes tipo2 y esteatosis hepática1. La HTG se define con concentraciones de triglicéridos (TG) superiores a 200mg/dl, y se considera grave cuando estos se encuentran por encima de 500mg/dl. El fenotipo de la HTG puede cursar con xantomas palmares, esplenomegalia, pancreatitis lipidémica y formación de la placa de ateroma que puede conllevar un aumento del evento cardiovascular2. La mayor parte de las HTG son consecuencia de factores exógenos como la obesidad, el consumo excesivo de alcohol, una diabetes mal controlada, enfermedad renal crónica o consumo de ciertos fármacos, como estrógenos o ácido retinoico, y se producen por aumento de la síntesis hepática de partículas VLDL, por defectos en el catabolismo periférico de quilomicrones y VLDL, o por defectos en la captación hepática de remanentes3,4.
Las formas primarias más graves de HTG se producen por defectos en el catabolismo periférico de las lipoproteínas ricas en TG. La lipoproteína lipasa (LpL) hidroliza los TG de quilomicrones y VLDL, dependiendo de la apolipoproteínaC-II como cofactor esencial para su activación. Recientemente se han descrito 2 proteínas: Lmf1 (factor de maduración de lipasas)5 y Gpihbp1, que intervienen en este proceso de hidrólisis, favoreciendo la dimerización de la LpL y su anclaje endotelial, respectivamente6,7
Se han descrito varias mutaciones en el gen LMF1 que producen HTG grave por defectos en la actividad LpL, lo que confirma el papel de LMF1 en la regulación del metabolismo de los TG5,8; sin embargo, se desconoce su frecuencia y su posible implicación en formas menos graves de HTG.
El presente trabajo planteó como objetivo identificar variantes génicas, comunes y no comunes, en el gen LMF1 en sujetos con HTG grave de causa primaria, analizar bioinformáticamente su efecto, y conocer si existe asociación entre las variantes alélicas de LMF1 y la HTG.
Material y métodosSujetos del estudioSe seleccionó un grupo de 112 sujetos consecutivos y no relacionados procedentes de la Unidad Clínica y de Investigación en Lípidos y Arteriosclerosis del Hospital Universitario Miguel Servet, con edades comprendidas entre los 21 y los 73años, afectos de HTG grave, definida por niveles de TG>500mg/dl sin tratamiento hipolipemiante en al menos 2 determinaciones diferentes, y al menos una de ellas tras una dieta baja en grasas.
Fueron criterios de exclusión las hipertrigliceridemias secundarias: diabetes descompensada (HbA1c≥7,5%), obesidad (índice de masa corporal [IMC] ≥30kg/m2), consumo de alcohol superior a 30g/día, insuficiencia renal crónica (filtrado glomerular por debajo de 30ml/min), hemocromatosis, hipotiroidismo y tratamiento con fármacos que aumentan los TG, como estrógenos, antirretrovirales, corticoides, andrógenos y ácido retinoico.
Todos los pacientes firmaron un consentimiento informado, que fue previamente aprobado por el comité ético local (Comité Ético de Investigación Clínica de Aragón).
Determinaciones clínicas y bioquímicasSe elaboró una historia clínica completa que incluyó: edad, antecedentes personales y familiares de hipertrigliceridemia y enfermedad cardiovascular, y sus diferentes factores de riesgo, consumo de tabaco, alcohol, nivel de actividad física y consumo de fármacos. La exploración clínica incluyó: presión arterial sistólica y diastólica, peso, talla, perímetro de cintura, presencia de arco corneal, xantomas tendinosos y xantelasmas. Se calculó el IMC y el consumo total de tabaco (número de paquetes de cigarrillos al día×número de años fumando).
Se extrajo una muestra de sangre tras, al menos, 10h de ayunas, sin tratamiento hipolipemiante en las últimas 5semanas, y se determinaron las concentraciones de: colesterol total, colesterol HDL (cHDL), TG, apoA-I, apoB, Lp(a), HbA1c, glucosa y TSH.
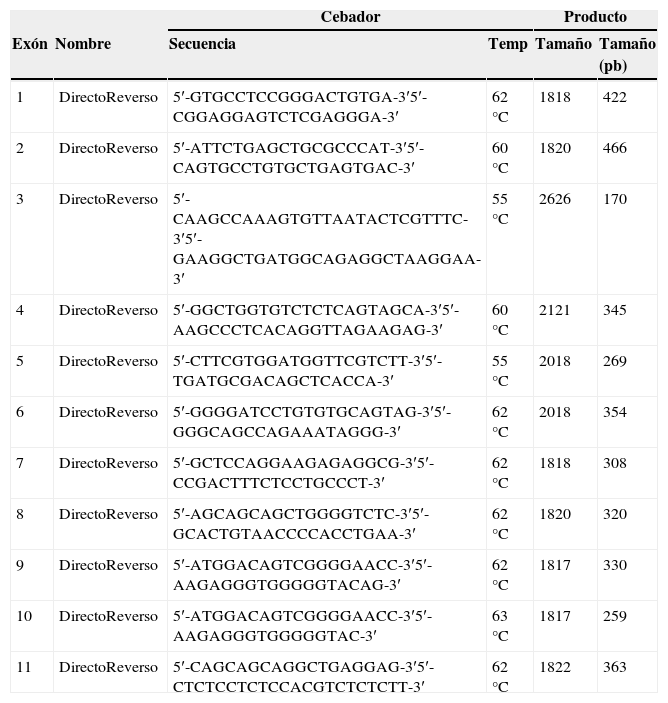
Análisis genéticoSe aisló el ADN de cada uno de los sujetos del estudio y se realizó la secuenciación del gen LMF1. Para ello, se amplificó por PCR el promotor, los 11 exones y las uniones exón-intrón, utilizando como secuencia de referencia NM_022773. Para la amplificación se usaron diferentes programas térmicos para cada exón (tabla 1).
Cebadores para la amplificación del gen LMF1
| Cebador | Producto | ||||
|---|---|---|---|---|---|
| Exón | Nombre | Secuencia | Temp | Tamaño | Tamaño (pb) |
| 1 | DirectoReverso | 5′-GTGCCTCCGGGACTGTGA-3′5′-CGGAGGAGTCTCGAGGGA-3′ | 62°C | 1818 | 422 |
| 2 | DirectoReverso | 5′-ATTCTGAGCTGCGCCCAT-3′5′-CAGTGCCTGTGCTGAGTGAC-3′ | 60°C | 1820 | 466 |
| 3 | DirectoReverso | 5′-CAAGCCAAAGTGTTAATACTCGTTTC-3′5′-GAAGGCTGATGGCAGAGGCTAAGGAA-3′ | 55°C | 2626 | 170 |
| 4 | DirectoReverso | 5′-GGCTGGTGTCTCTCAGTAGCA-3′5′-AAGCCCTCACAGGTTAGAAGAG-3′ | 60°C | 2121 | 345 |
| 5 | DirectoReverso | 5′-CTTCGTGGATGGTTCGTCTT-3′5′-TGATGCGACAGCTCACCA-3′ | 55°C | 2018 | 269 |
| 6 | DirectoReverso | 5′-GGGGATCCTGTGTGCAGTAG-3′5′-GGGCAGCCAGAAATAGGG-3′ | 62°C | 2018 | 354 |
| 7 | DirectoReverso | 5′-GCTCCAGGAAGAGAGGCG-3′5′-CCGACTTTCTCCTGCCCT-3′ | 62°C | 1818 | 308 |
| 8 | DirectoReverso | 5′-AGCAGCAGCTGGGGTCTC-3′5′-GCACTGTAACCCCACCTGAA-3′ | 62°C | 1820 | 320 |
| 9 | DirectoReverso | 5′-ATGGACAGTCGGGGAACC-3′5′-AAGAGGGTGGGGGTACAG-3′ | 62°C | 1817 | 330 |
| 10 | DirectoReverso | 5′-ATGGACAGTCGGGGAACC-3′5′-AAGAGGGTGGGGGTAC-3′ | 63°C | 1817 | 259 |
| 11 | DirectoReverso | 5′-CAGCAGCAGGCTGAGGAG-3′5′-CTCTCCTCTCCACGTCTCTCTT-3′ | 62°C | 1822 | 363 |
Los cebadores de LMF1 utilizados se basan en los descritos por Peterfy et al.5
Para cada reacción de amplificación se mezcló la enzima BioTaq DNA polimerasa (Bioline), tampón 10x, MgCl2, dNTPs, cebadores correspondientes (que se muestran en la tabla 1tabla 1), el ADN de interés y agua destilada estéril. El resultado de las diferentes amplificaciones se comprobó mediante electroforesis en gel de agarosa 2% en TAE1X teñido con SYBR® Safe. Para la purificación de los productos de PCR se utilizó ExoSAP-IT® (USB). Se preparó una dilución 1:10 de ExoSAP-IT® (USB) en dH2O, de la que se añadieron 2μl a 5μl del producto de PCR, incubando a 37°C durante 45min, y a continuación 80°C durante a 15min. Se secuenciaron los fragmentos purificados utilizando el producto comercial BigDye® Terminator v3.1 CycleSequencing Kit (AppliedBiosystems). Se desnaturalizaron los productos de la secuenciación mediante una incubación durante 2min a 94°C. La electroforesis capilar se llevó a cabo en el secuenciador automático ABI 3500XL (AppliedBiosystems). El alineamiento y la lectura de las secuencias obtenidas se llevaron a cabo mediante el programa informático VariantReporter™ (AppliedBiosystems).
Análisis bioinformáticoPara evaluar la funcionalidad de las variantes génicas identificadas se usaron los siguientes programas informáticos: PolyPhen-29, SIFT10 y MutationT@ster11.
PolyPhen-2 clasificó las variantes en benignas, posiblemente dañinas y probablemente dañinas, de acuerdo a la conservación de la secuencia y la estructura de la proteína. SIFT clasificó las variantes en función de la homología de secuencia, como tolerante o intolerante. MutationT@ster se utilizó también para variantes que incluían codones de parada y alteraciones intrónicas o complejas, clasificándolas como posiblemente causales de la enfermedad o posiblemente polimorfismos. El efecto de las variantes en sitios potenciales de ayuste se estudió mediante NetGene212 y NNSplice13, analizando la estructura de los sitios donador y aceptor.
Análisis estadísticoSe realizó con el software SPSS versión 20.0 (Chicago, Ilinois, EE.UU.) usando un nivel de significación p<0,05. Para conocer si las variables presentaron una distribución normal se realizó un análisis de Kolmogorov-Smirnov. Los datos se expresaron como media±desviación estándar para las variables numéricas con una distribución normal y se analizaron con la prueba t de Student, mientras que aquellas que no siguieron distribución normal se expresaron como mediana y rango intercuartil y se analizaron con el test U de Mann-Whitney. Las variables cualitativas, incluyendo la comparación de frecuencias alélicas y genotípicas, se expresaron en porcentaje y se analizaron mediante el test Chi-cuadrado; dichas frecuencias se compararon con los datos obtenidos del proyecto 1000 Genomas en el subestudio realizado con población general europea14.
Se realizó un estudio de regresión logística lineal multivariante, introduciendo como variables independientes: edad, sexo, perímetro de cintura, presencia de diabetes y todas las variantes génicas identificadas en este estudio.
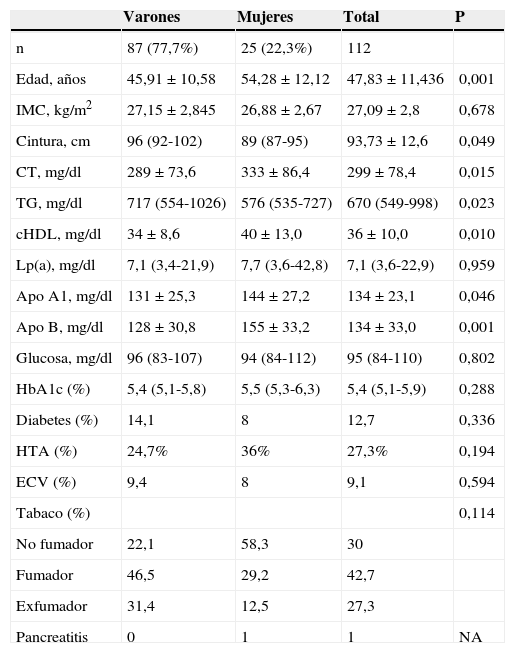
ResultadosCaracterísticas de los sujetosEl estudio incluyó 112 sujetos: 87 varones y 25 mujeres. En la tabla 2 se muestran las características clínicas y bioquímicas de los sujetos estudiados. La media de edad fue 47,8, y la mediana de concentración de TG fue de 670mg/dl. Las mujeres, en comparación con los varones, eran mayores, con concentraciones más elevadas de colesterol, cHDL, apoA-I y apoB (p<0,05) y menor concentración de TG (p<0,05).
Características de la población de estudio
| Varones | Mujeres | Total | P | |
|---|---|---|---|---|
| n | 87 (77,7%) | 25 (22,3%) | 112 | |
| Edad, años | 45,91±10,58 | 54,28±12,12 | 47,83±11,436 | 0,001 |
| IMC, kg/m2 | 27,15±2,845 | 26,88±2,67 | 27,09±2,8 | 0,678 |
| Cintura, cm | 96 (92-102) | 89 (87-95) | 93,73±12,6 | 0,049 |
| CT, mg/dl | 289±73,6 | 333±86,4 | 299±78,4 | 0,015 |
| TG, mg/dl | 717 (554-1026) | 576 (535-727) | 670 (549-998) | 0,023 |
| cHDL, mg/dl | 34±8,6 | 40±13,0 | 36±10,0 | 0,010 |
| Lp(a), mg/dl | 7,1 (3,4-21,9) | 7,7 (3,6-42,8) | 7,1 (3,6-22,9) | 0,959 |
| Apo A1, mg/dl | 131±25,3 | 144±27,2 | 134±23,1 | 0,046 |
| Apo B, mg/dl | 128±30,8 | 155±33,2 | 134±33,0 | 0,001 |
| Glucosa, mg/dl | 96 (83-107) | 94 (84-112) | 95 (84-110) | 0,802 |
| HbA1c (%) | 5,4 (5,1-5,8) | 5,5 (5,3-6,3) | 5,4 (5,1-5,9) | 0,288 |
| Diabetes (%) | 14,1 | 8 | 12,7 | 0,336 |
| HTA (%) | 24,7% | 36% | 27,3% | 0,194 |
| ECV (%) | 9,4 | 8 | 9,1 | 0,594 |
| Tabaco (%) | 0,114 | |||
| No fumador | 22,1 | 58,3 | 30 | |
| Fumador | 46,5 | 29,2 | 42,7 | |
| Exfumador | 31,4 | 12,5 | 27,3 | |
| Pancreatitis | 0 | 1 | 1 | NA |
Apo A1: apolipoproteína A1; Apo B: apolipoproteínaB; cHDL: colesterol HDL; CT: colesterol total; ECV: enfermedad cardiovascular; HTA: hipertensión arterial; IMC: índice de masa corporal; Lp(a): lipoproteína (a); NA: no aplica; TG: triglicéridos.
Los datos de variables cuantitativas se expresan como media±desviación estándar, excepto para las variables que no siguen la distribución normal (mediana, rango intercuartil). Las variables cualitativas se expresan como porcentaje (%).
El valor p fue calculado por las pruebas t de Student, U de Mann-Whitney o Chi-cuadrado, según fuera conveniente.
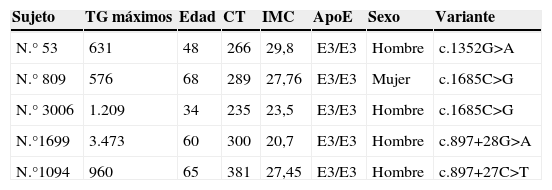
En la población estudiada se identificaron las variantes raras c.1352G>A (p.Arg451Trp), c.897+28G>A, c.897+27C>T y c.1685C>G (p.Pro562Arg) que se muestran en la tabla 3, considerando como tales aquellas que no habían sido encontradas en el proyecto de los 1000 Genomas.
Características clínicas de los sujetos portadores de las variantes raras del gen LMF1
| Sujeto | TG máximos | Edad | CT | IMC | ApoE | Sexo | Variante |
|---|---|---|---|---|---|---|---|
| N.° 53 | 631 | 48 | 266 | 29,8 | E3/E3 | Hombre | c.1352G>A |
| N.° 809 | 576 | 68 | 289 | 27,76 | E3/E3 | Mujer | c.1685C>G |
| N.° 3006 | 1.209 | 34 | 235 | 23,5 | E3/E3 | Hombre | c.1685C>G |
| N.°1699 | 3.473 | 60 | 300 | 20,7 | E3/E3 | Hombre | c.897+28G>A |
| N.°1094 | 960 | 65 | 381 | 27,45 | E3/E3 | Hombre | c.897+27C>T |
CT: colesterol total; IMC: índice de masa corporal; TG: triglicéridos.
Los TG y el CT están expresados en mg/dl.
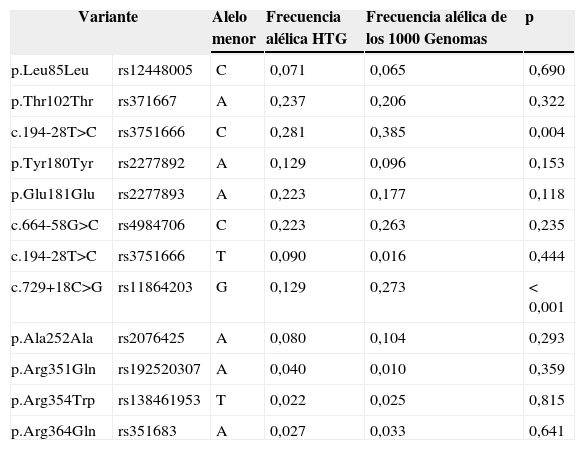
Las variantes comunes en el gen LMF1, y sus frecuencias alélicas, se muestran en la tabla 4. Los polimorfismos c.194-28T>C y c.729+18C>G mostraron una frecuencia significativamente menor en los sujetos con HTG en comparación con la población europea del estudio de los 1000 Genomas (p<0,05).
Frecuencias alélicas de los SNP identificados en el gen LMF1 en la población HTG estudiada y los 1000 Genomas de la población europea
| Variante | Alelo menor | Frecuencia alélica HTG | Frecuencia alélica de los 1000 Genomas | p | |
|---|---|---|---|---|---|
| p.Leu85Leu | rs12448005 | C | 0,071 | 0,065 | 0,690 |
| p.Thr102Thr | rs371667 | A | 0,237 | 0,206 | 0,322 |
| c.194-28T>C | rs3751666 | C | 0,281 | 0,385 | 0,004 |
| p.Tyr180Tyr | rs2277892 | A | 0,129 | 0,096 | 0,153 |
| p.Glu181Glu | rs2277893 | A | 0,223 | 0,177 | 0,118 |
| c.664-58G>C | rs4984706 | C | 0,223 | 0,263 | 0,235 |
| c.194-28T>C | rs3751666 | T | 0,090 | 0,016 | 0,444 |
| c.729+18C>G | rs11864203 | G | 0,129 | 0,273 | < 0,001 |
| p.Ala252Ala | rs2076425 | A | 0,080 | 0,104 | 0,293 |
| p.Arg351Gln | rs192520307 | A | 0,040 | 0,010 | 0,359 |
| p.Arg354Trp | rs138461953 | T | 0,022 | 0,025 | 0,815 |
| p.Arg364Gln | rs351683 | A | 0,027 | 0,033 | 0,641 |
Todas las variantes se describen de acuerdo con las últimas recomendaciones de HGVS (http://www.hgvs.org/mutnomen). El valor p fue calculado por el test χ2.
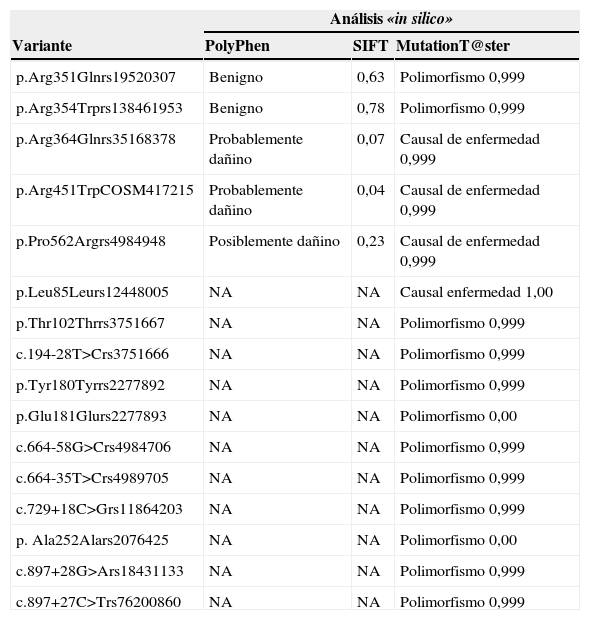
Todas las variantes que supusieron un cambio de aminoácido no sinónimo se analizaron con los programas informáticos PolyPhen-29, SIFT10 y MutationT@ster11. Las mutaciones que se encontraban en el promotor o dieron lugar a un codón de parada solo pudieron ser analizadas por MutationT@ster. El resultado de este análisis bionformático de las variantes del gen LMF1 se muestra en la tabla 5.
Análisis bioinformático de las variantes encontradas en el gen LMF1
| Análisis «in silico» | |||
|---|---|---|---|
| Variante | PolyPhen | SIFT | MutationT@ster |
| p.Arg351Glnrs19520307 | Benigno | 0,63 | Polimorfismo 0,999 |
| p.Arg354Trprs138461953 | Benigno | 0,78 | Polimorfismo 0,999 |
| p.Arg364Glnrs35168378 | Probablemente dañino | 0,07 | Causal de enfermedad 0,999 |
| p.Arg451TrpCOSM417215 | Probablemente dañino | 0,04 | Causal de enfermedad 0,999 |
| p.Pro562Argrs4984948 | Posiblemente dañino | 0,23 | Causal de enfermedad 0,999 |
| p.Leu85Leurs12448005 | NA | NA | Causal enfermedad 1,00 |
| p.Thr102Thrrs3751667 | NA | NA | Polimorfismo 0,999 |
| c.194-28T>Crs3751666 | NA | NA | Polimorfismo 0,999 |
| p.Tyr180Tyrrs2277892 | NA | NA | Polimorfismo 0,999 |
| p.Glu181Glurs2277893 | NA | NA | Polimorfismo 0,00 |
| c.664-58G>Crs4984706 | NA | NA | Polimorfismo 0,999 |
| c.664-35T>Crs4989705 | NA | NA | Polimorfismo 0,999 |
| c.729+18C>Grs11864203 | NA | NA | Polimorfismo 0,999 |
| p. Ala252Alars2076425 | NA | NA | Polimorfismo 0,00 |
| c.897+28G>Ars18431133 | NA | NA | Polimorfismo 0,999 |
| c.897+27C>Trs76200860 | NA | NA | Polimorfismo 0,999 |
NA: no analizado.
Todas las variantes se describen de acuerdo con las últimas recomendaciones de HGVS (http://www.hgvs.org/mutnomen). Las predicciones de los efectos deletéreos se realizaron utilizando SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) y MutationT@ster (http://www.mutationtaster.org/).
El análisis mediante PolyPhen-2, MutationT@ster y SIFT mostró que las variantes p.Arg364Gln, p.Arg451Trp y p.Pro562Arg eran potencialmente dañinas y posiblemente la causa de la HTG que presentan los sujetos portadores.
Modelo de regresión logística lineal entre las variables génicas encontradas y la concentración de triglicéridosAl realizar el análisis de regresión lineal entre todas las variantes encontradas en el gen LMF1 y la concentración de TG, se encontró que la variante p.Leu85Leu explicaba el 11,7% de la variabilidad en la concentración de TG, ajustada por edad, sexo, perímetro de cintura y presencia de diabetes de manera estadísticamente significativa (tabla 6).
Variables independientemente asociadas con la concentración de TG mediante regresión logística lineal
| Variable | Coeficiente tipificado beta | p | R2 corregida |
|---|---|---|---|
| p.Leu85Leu | −0,265 | 0,005 | 0,117 |
Modelo de regresión logística lineal para TG (1/TG) como variable dependiente. El modelo ha sido ajustado por edad, sexo, perímetro de cintura y presencia de diabetes.
LMF1 codifica una proteína de membrana localizada en el retículo endoplasmático, que es esencial para la maduración tanto de LpL como de la lipasa hepática. La forma activa de LpL actúa como un homodímero, y Lmf1 favorece su dimerización15,16. Por tanto, mutaciones que generen una pérdida de función en LMF1 es de esperar que afecten a la actividad de LpL, como ya ha sido demostrado anteriormente y confirman nuestros resultados5,8.
En este trabajo hemos identificado 5 pacientes (4,46% de 112) portadores de 4 variantes raras en heterocigosidad, c.1352G>A (p.Arg451Trp), c.897+28G>A, c.897+27C>T y c.1685C>G (p.Pro562Arg) en el gen LMF1 asociadas previamente a HTG. Además, hemos identificado variantes comunes o polimorfismos que se asocian con la HTG, ya que se encuentran con una frecuencia alélica diferente de la que se observa en población general: c.194-28T>G y c.729+18C>G. Los pacientes de este estudio presentaron una concentración media de TG de 670 mg/dl, lo que explica la menor frecuencia de defectos raros (4,46%) en nuestro estudio comparada con el trabajo publicado por Surendran et al.20, quienes encontraron 8 pacientes (9,3%) portadores de variantes raras en el LMF1 seleccionados por sufrir hiperquilomicronemia y, por tanto, con niveles más altos de TG.
Varias conclusiones, a nuestro juicio importantes, se derivan de nuestros resultados.
En primer lugar, defectos genéticos funcionalmente importantes en LMF1 en heterocigosidad se asocian con HTG en un porcentaje pequeño pero significativo.
LMF1 se ha asociado recientemente con niveles elevados de TG cuando hay mutaciones en homocigosidad en el dominio C-terminal de la proteína10,15. Las variantes raras de LMF1 p.Arg451Trp y p.Pro562Arg son dañinas de acuerdo con el análisis bioinformático y se encuentran localizadas cercanas a la región crítica para la dimerización de LpL17. Para determinar si los polimorfismo c.194-28T>G y c.729+18C>G contribuyen a la causa de HTG en los sujetos portadores harían falta más estudios tanto epidemiológicos como funcionales.
De los 5 pacientes que son portadores de las variantes raras, todos presentan TG máximos entre 576 y 3.473mg/dl, con una mediana de 1.370mg/dl. Destacan 2 de los pacientes, uno con 60años y TG de 3.473mg/dl portador de la variante c.897+28 G>A con tan solo un IMC de 20,7kg/m2, y otro portador de la variante c.1685C>G de 23años y con 23,5kg/m2 de IMC. Por tanto, parece indicar que estas mutaciones ejercen un efecto importante en la función de LMF1. Si estos sujetos tienen asociadas otras mutaciones en otros genes desconocidos que han contribuido junto con LMF1 en el desarrollo de la HTG, no podemos descartarlo. Los sujetos portadores de mutaciones graves en heterocigosidad en genes causantes de hipertrigliceridemia tienen concentraciones de TG muy variables, desde la normalidad a la HTG grave, lo que pone de manifiesto la complejidad de la etiología de la HTG, y la posibilidad de otros factores asociados18.
En segundo lugar, diferencias en la frecuencia de determinados alelos en LMF1 apoyan la idea que la variación genética en LMF1 contribuye a la patogenia de la HTG. Recientemente ha sido demostrado que los pacientes con HTG muestran un exceso de variantes raras en los genes seleccionados por los GWAS (APOA5, GCKR, LPL y APOB) comparados con la población normolipémica, y que, por tanto, muchas HTG son el resultado de la acumulación de variaciones genéticas comunes con efectos hipertrigliceridémicos pequeños de forma aislada, pero confiriendo susceptibilidad especialmente si la acumulación es elevada, o se combinan con otras situaciones ambientales. Sin embargo, las variantes comunes de estos genes tan solo explicarían en un 25% las diferencias de las concentraciones lipídicas en plasma, y esto sugiere el potencial genético que queda por dilucidar en el campo de la HTG. Ante esta situación se han realizado diferentes estudios intentando aumentar el background genético de esta patología, estudiando genes como APOC2, GPIHBP1 y LMF1, entre otros, donde ha quedado demostrada la implicación de estos genes en la patología de la HTG17,19.
Por último, nuestros resultados confirman que la HTG primaria es heterogénea en su etiología y que posiblemente se produce por la acumulación de variantes comunes y raras en los genes involucrados en el metabolismo de los TG.
En conclusión, nuestros resultados sugieren que la HTG primaria del adulto con concentración de TG superior a 500mg/dl presenta mayor cantidad de variantes genéticas en el gen LMF1 respecto a la población general, con una combinación de mutaciones de efecto entre moderado y agresivo, y polimorfismos clásicamente asociados con esta dislipidemia.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciaciónTrabajo financiado con el proyecto PI12/01087 y la RETIC RD12/0042/0055.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.