






La resistencia a la insulina precede y predice la presencia de diabetes mellitus tipo2, condición que supone un notable incremento del riesgo cardiovascular. La interleucina-6 es uno de los mediadores que relacionan la inflamación crónica observada en estados de obesidad con la resistencia a la insulina a través de la activación de STAT3 (signal transducer and activator of transcription 3), con el consiguiente aumento de SOCS3 (suppressor of cytokine signaling 3) en el hígado. El objetivo de este estudio ha sido evaluar si un agonista del receptor activado por proliferadores peroxisómicos (PPAR) β/δ, GW501516, es capaz de evitar la activación de la vía de señalización IL-6/STAT3/SOCS3 y la resistencia a la insulina en células hepáticas.
Material y métodosCélulas HepG2 humanas se estimularon con IL-6 (20ng/ml) en presencia o en ausencia de GW501516 (10μM). También analizamos el hígado de ratones salvajes y con deficiencia PPARβ/δ. Los niveles de ARNm y proteínas se analizaron mediante las técnicas de RT-PCR y Western-Blot, respectivamente.
ResultadosGW501516 evitó la fosforilación en Tyr705 y en Ser727 de STAT3 y el aumento de SOCS3 inducidas por la IL-6. Asimismo, el tratamiento con este fármaco evitó la activación por la IL-6 de la ERK1/2, una serina-treonina cinasa implicada en la fosforilación de STAT3 en Ser727. Cabe destacar que el hígado de ratones deficientes en PPARβ/δ mostró un aumento de la fosforilación en Tyr705 y Ser727 de STAT3 así como un nivel más alto de fosforilación de ERK1/2 respecto a los observados en ratones salvajes. Finalmente, estos cambios podrían explicar por qué GW501516 mejoró la fosforilación de la Akt inducida por insulina y los niveles de proteína de IRS-1 e IRS-2 en presencia de IL-6 en células HepG2.
ConclusionesEstos resultados indican que el agonista de PPARβ/δ, GW501516, es capaz de evitar la activación de STAT3 inducida por IL-6 mediante la inhibición de la fosforilación de ERK1/2. Estos efectos podrían contribuir a mejorar la resistencia a la insulina inducida por citocinas en células hepáticas.
Insulin resistance precedes and predicts the development of type 2 diabetes mellitus, a disease which increases the risk of cardiovascular events. Interleukin (IL)-6 is one of the mediators linking obesity-derived chronic inflammation with insulin resistance through activation of STAT3, with subsequent upregulation of suppressor of cytokine signaling 3 (SOCS3) in liver. The aim of this study was to evaluate whether peroxisome proliferator-activated receptor (PPAR) β/δ agonist GW501516 prevented activation of the IL-6/STAT3/SOCS3 pathway and insulin resistance in hepatic cells.
Material and methodsHuman HepG2 cells were stimulated for 10min with IL-6 (20ng/mL) in the presence or in the absence of 10μM GW501516, then mRNA and protein levels were analyzed by RT-PCR or Western-Blot, respectively. In addition, we also analyzed protein levels from PPARβ/δ null mice and wild-type mice livers.
ResultsGW501516 prevented IL-6-induced STAT3 phosphorylation on Tyr705 and Ser727 and avoided the increase in SOCS3 caused by this cytokine. In addition, this drug also prevented IL-6-dependent ERK1/2 phosphorylation, a serine-threonine protein kinase involved in STAT3 phosphorylation on Ser727. Interestingly, livers from PPARβ/δ null mice showed increased phosphorylations on Tyr 705 and Ser727 of STAT3 as well as phosphorylated ERK1/2 levels. Finally, all these changes might contribute to explain why GW501516 improved the reductions in insulin-stimulated Akt phosphorylation and in IRS-1 and IRS-2 protein levels caused by IL-6 in hepatic cells.
ConclusionsThese findings indicate that the PPARβ/δ agonist GW501516 prevents IL-6-induced STAT3 activation by inhibiting ERK1/2 phosphorylation. These effects of GW501516 may contribute to the prevention of cytokine-induced insulin resistance in hepatic cells.
La resistencia a la insulina (RI) precede y predice la presencia de diabetes mellitus tipo2 (DM2), condición que supone un notable incremento del riesgo cardiovascular1. Asimismo, un gran número de evidencias sugieren que tanto la RI como la DM2 se asocian con un estado inflamatorio crónico de baja intensidad. Además, la RI se correlaciona con un aumento de los niveles de los marcadores de respuesta a la fase aguda como el factor de necrosis tumoral alfa (TNF-α)2,3, la interleucina (IL)-1β3 y la IL-64,5. De estas citocinas, la IL-6 muestra una mayor asociación con la obesidad tanto en modelos de ratones como en modelos humanos. Así pues, en humanos se observa que niveles altos de IL-6 se correlacionan directamente con obesidad y RI y predicen el desarrollo de DM26-8, mientras que la supresión de IL-6 mejora la acción de la insulina en un modelo de obesidad en ratones9. In vitro, la IL-6 induce RI en células hepáticas10,11.
La señalización de la IL-6 se inicia con la unión de esta citocina a su complejo receptor de membrana, el cual contiene la glucoproteína gp130, que activa diferentes Janus tyrosine kinases (Jak1, Jak2, Tyk2), con la consiguiente fosforilación de la Tyr705 de STAT3 (signal transducer and activator of transcription 3)12. Este STAT3 fosforilado dimeriza y transloca al núcleo, donde regula la transcripción de sus genes dianas mediante la unión específica a sus elementos de respuesta en el ADN13. Además de la activación por la fosforilación en la Tyr705, STAT3 también necesita ser fosforilado en la Ser727 para adquirir su máxima actividad transcripcional14,15. Entre las cinasas involucradas en esta fosforilación se incluyen la proteincinasa C (PKC), la Jnk (Jun N-terminal kinase), la ERK (extracellular signal-related kinase), la cinasa activada por mitógeno p38 y la Mtor (mammalian target of rapamycin)16.
Se ha descrito que la IL-6 induce RI en el hígado a través de la activación de STAT3 y la consiguiente inducción de la proteína supresora de la señalización de citocinas 3 (SOCS3, suppressor of cytokine signaling 3)9,17,18. Las proteínas SOCS fueron originalmente descritas como moléculas inducibles por citocinas involucradas en un feedback negativo entre citocinas19 y la señalización de la insulina20. Diferentes estudios han descrito que SOCS3 puede inhibir la señalización de la insulina20-22 mediante la interacción directa con el receptor de la insulina y evitando la unión del sustrato del receptor de la insulina (insulin receptor substrate 1, IRS-1) con el receptor de la insulina, y por lo tanto, inhibiendo la fosforilación en tirosina de IRS-1 y la siguiente cascada de fosforilaciones downstream20,21. Además, SOCS3 inhibe la señalización de la insulina a través de la degradación proteasomal de IRS-122,23. En el hígado, la sobreexprexión de SOCS3 provoca RI, mientras que la supresión de SOCS3 en ratones obesos y diabéticos (db/db) atenúa la RI24.
Los receptores activados por proliferadores peroxisómicos (PPAR) son miembros de la superfamilia de los factores de transcripción inducibles por ligando que forman heterodímeros con receptores retinoides X (RXR) y que se unen a lugares consensos del ADN25. Además, los PPAR pueden suprimir la inflamación mediante diferentes mecanismos, como la reducción de la liberación de factores inflamatorios o la estabilización de complejos represores en los promotores de genes inflamatorios26. De las 3 isoformas de PPAR descritas en mamíferos, PPARα (también conocido como NR1C1) y PPARγ (NR1C3) son diana para fármacos hipolipemiantes (fibratos) y antidiabéticos (tiazolidindionas o glitazonas), respectivamente. Finalmente, la activación del tercer isotipo, PPARβ/δ (NR1C2), aumenta el catabolismo de los ácidos grasos en el tejido adiposo y en el músculo esquelético, y por eso esta isoforma se ha propuesto como un tratamiento potencial para la RI27. Recientemente se ha publicado que la activación de PPARβ/δ interfiere en la reacción de fase aguda en el hígado inhibiendo la actividad transcripcional de STAT328, aunque todavía se desconoce el mecanismo implicado.
Teniendo en cuenta que en la RI mediada por IL-6 en el hígado la vía de señalización STAT3-SOCS3 juega un papel importante, el objetivo de este estudio fue evaluar si la activación de PPARβ/δ por GW501516 evitaba la RI mediada por la IL-6 en células hepáticas humanas y los mecanismos implicados. El tratamiento con el agonista de PPARβ/δ, GW501516, evitó la inducción mediada por IL-6 de los niveles de ARNm de SOCS3 y de los niveles proteicos de STAT3 fosforilada en Tyr705 y Ser727 en células HepG2. Además, la exposición a IL-6 provocó un aumento de los niveles de ERK1/2 fosforilada, pero este aumento no se observó en presencia de GW501516. En concordancia con la capacidad de PPARβ/δ para bloquear la actividad de STAT3 inducida por IL-6, los hígados de ratones deficientes en PPARβ/δ mostraron niveles de fosforilación de STAT3 en Tyr705 y Ser727 más elevados que los que presentaban los ratones salvajes. De acuerdo con la inhibición de la vía de señalización STAT3-SOCS3 causada por GW501516, este fármaco también evitó la reducción inducida por IL-6 de los niveles de AKT fosforilada estimulada con insulina, así como la reducción de los niveles proteicos de IRS-1 e IRS-2. Todos estos datos sugieren que la activación de PPARβ/δ por GW501516 en hepatocitos humanos evita la activación de la vía STAT3-SOCS3 inducida por IL-6 a través de la inhibición de ERK1/2. Este efecto de PPARβ/δ podría contribuir a reducir la RI en hepatocitos humanos.
MétodosEl ligando de PPARδ GW501516 se obtuvo de Biomol Research Labs Inc. (Plymouth Meeting, Pensilvania). Otros reactivos procedían de Sigma (St. Louis, Misuri).
Cultivo celularLas células HepG2 (carcinoma hepatocelular, American Type Culture Collection, Manassas, VA, EE.UU.) se mantuvieron en Dulbecco's Modified Eagle's Medium (DMEM) (Lonza, Barcelona, España) que incluye 4,5g/l de glucosa y L-glutamina, suplementado con un 10% (v/v) de FBS (Invitrogen, San Diego, CA, EE.UU.), penicilina-estreptomicina (Invitrogen) y aminoácidos no esenciales. La densidad celular se ajustó a 2×105 células/ml y se añadió por cada pozo de una placa de 12 pocillos 1ml de la suspensión celular (NUNC, Roskilde, Dinamarca). Entonces las células HepG2 se incubaron con GW501516 (10μM) e IL-6 (20ng/ml) a los tiempos indicados. Después de la incubación, el ARN y los extractos de proteína total y nuclear se extrajeron como se describe posteriormente. Los inhibidores se añadieron 30min antes de la incubación con IL-6.
AnimalesLa generación de los ratones deficientes para PPARβ/δ se realizó tal y como se describió previamente29. Se utilizaron 6 machos deficientes para PPARβ/δ y 6 machos control «salvajes» de 5 a 6 meses de edad. De acuerdo con las normas especificadas en la oficina de veterinaria de Lausana (Suiza), los ratones se mantuvieron bajo un ciclo de luz-oscuridad estándar (12h luz/oscuridad) a una temperatura de 21±1°C y se alimentaron con pienso Provimi Kliba. Se extrajo tejido hepático, que inmediatamente fue congelado en nitrógeno líquido y posteriormente almacenado a –80°C.
Análisis de los niveles de ARNmLos niveles de ARN se estudiaron mediante la reacción en cadena de la polimerasa de transcripción reversa (RT-PCR), tal como se ha descrito previamente30. El ARN total se aisló utilizando el reactivo Ultraspec (Biotecx, Houston). El ARN total aislado mediante este método no está degradado y está libre de contaminaciones por ADN o proteínas. Las secuencias de los primers sentido y antisentido utilizados para la amplificación fueron: SOCS3 (Suppressor of cytokine signaling 3) 5’-TTTTCGCTGCAGAGTGACCCC-3’ and 5’-TGGAGGAGAGAGGTCGGCTCA-3’; y 18S, 5’-ATGACTTCCAAGCTGGCCGTG-3’ y 5’-GCGCAGTGTGGTCCACTCTCA-3’. La amplificación de cada gen muestra una única banda del tamaño esperado (SOCS3: 250bp y 18S: 333bp). Se llevaron a cabo experimentos preliminares para cada uno de los genes estudiados con varias cantidades de ADNc para determinar las condiciones no saturantes de la amplificación por PCR. Entonces, bajo estas condiciones, se evaluó la cuantificación relativa del ARNm por el método RT-PCR utilizado en este estudio31. Las bandas radiactivas se cuantificaron por escáner vídeo-densitométrico (Vilbert Lourmat Imaging). Los resultados para la expresión de los ARNm específicos siempre se presentan en relación con la expresión del gen control (18S).
Aislamiento de extractos nuclearesLos extractos nucleares se extrajeron tal como se ha descrito previamente32. Las placas de cultivo celular se rascaron con 1,5ml de tampón fosfato salino frío, se centrifugaron durante 10s y se resuspendieron los pellets en 400μl de tampón A frío (HEPES 10mM pH 7,9 a 4°C, MgCl2 1,5mM, KCl 10mM, DTT 0,5mM, PMSF 0,2mM y aprotenina 5μg/ml) mediante la inversión del tubo. Se dejaron las células durante 10min en hielo y después se sometieron a un vórtex durante 10s. Seguidamente se centrifugaron las muestras durante 10s y se descartó el sobrenadante. Los pellets se resuspendieron en 50μl de tampón C frío (HEPES-KOH 20mM, pH 7,9 a 4°C, 25% de glicerol, NaCl 420mM, MgCl2 1,5mM, EDTA 0,2mM, DTT 0,5mM, PMSF 0,2mM, aprotenina 5μg/ml y leupeptina 2μg/ml) y se incubaron durante 20min en hielo. Los restos celulares se eliminaron con una centrífuga de 2min a 4°C y la fracción del sobrenadante (que contiene proteínas de unión al ADN) se almacenó a –80°C. La concentración de extractos nucleares se determinó por la técnica de Bradford.
Western-blotLos anticuerpos contra la Akt total y fosforilada (Ser473), ERK1/2 fosforilada (Thr202/Tyr204) y STAT3 fosforilada (Tyr705 y Ser727) se adquirieron de Cell Signaling (Danvers, MA, EE.UU.). El anticuerpo contra STAT3 total se obtuvo de Santa Cruz (Santa Cruz, California, EE.UU.).
Para obtener proteína total, los hígados se homogeneizaron con tampón RIPA (Sigma) con inhibidores de proteasas y fosfatasas (PMSF, 0,2mM, ortovanadato de sodio 1mM y aprotenina 5,4μg/ml). El homogenado se centrifugó a 16.700g durante 30min a 4°C. La concentración de proteína se determinó mediante la técnica de Bradford.
Las proteínas obtenidas por este método, así como los extractos nucleares, se separaron por SDS-PAGE, se transfirieron a una membrana immobilon-poliviniliden-difluorada (Millipore, Bedford, MA, EE.UU.) y se incubaron con diferentes anticuerpos (como se muestra en los resultados). La detección de estos anticuerpos se consiguió mediante el kit de quimioluminiscencia EZ-ECL (Amersham, Piscataway, NJ, EE.UU.). El tamaño de las proteínas detectadas se estimó a partir de marcadores estándar de peso molecular (Invitrogen).
Análisis estadísticoLos datos se presentan como la media±desviación estándar de 5 experimentos separados. Las diferencias significativas se establecieron por ANOVA de una vía, utilizando el programa GraphPad Instat (Software GraphPad V2.03) (GraphPad Software, CA, EE.UU.). Cuando se encontraron variaciones significativas se aplicó el test de comparación múltiple de Tukey-Kramer. Las diferencias se consideraron significativas con un valor de p<0,05.
ResultadosPPARβ/δ evita la activación de la vía STAT3-SOCS3 inducida por interleucina-6 en hepatocitos humanosSe ha descrito que la activación de la vía de señalización STAT3-SOCS3 inducida por IL-6 en el hígado es en parte responsable del desarrollo de la resistencia a la insulina33, por eso decidimos estudiar si la activación de PPARβ/δ por GW501516 podría prevenir este efecto.
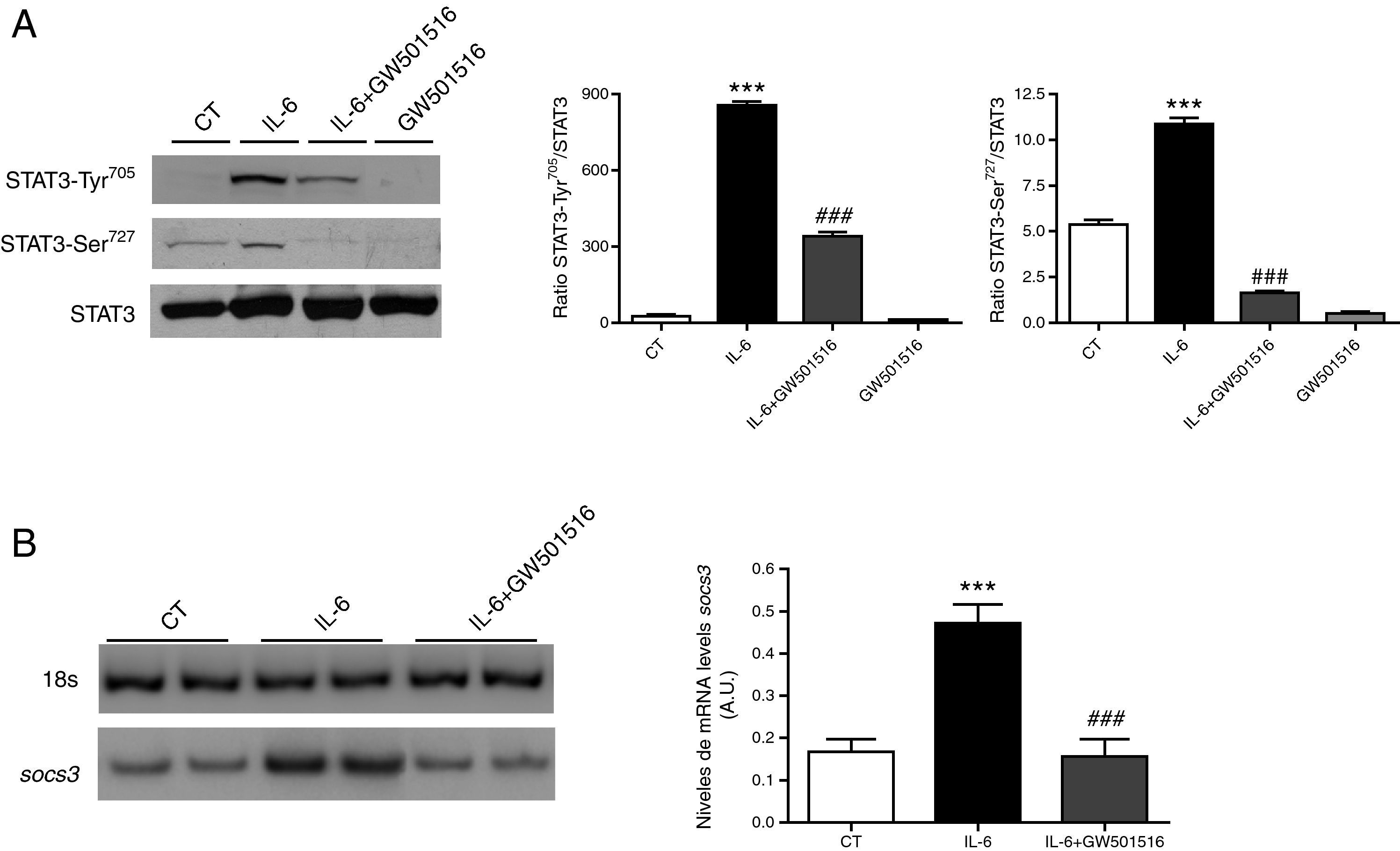
En primer lugar, estudiamos el estado de fosforilación de STAT3. La dimerización, la translocación al núcleo y el aumento en la actividad transcripcional de STAT3 requieren que este se fosforile en el residuo Tyr705. De acuerdo con esto, la exposición a IL-6 aumentó la fosforilación en Tyr705 de STAT3, pero la coincubación con GW501516 a 10μmol/l disminuyó este incremento (fig. 1A). Además, la fosforilación en el residuo de Ser727 de STAT3 es necesaria para que este adquiera su máxima actividad transcripcional14,15. Como se esperaba, la estimulación con IL-6 aumentó la fosforilación en este residuo, mientras que la presencia de GW501516 lo evitó (fig. 1A). Seguidamente estudiamos el efecto de la activación de PPARβ/δ sobre los niveles de ARNm del gen diana de STAT3, SOCS3. La presencia de IL-6 en el medio de cultivo provocó un aumento de los niveles de ARNm de SOCS3 (inducción de 2,7 veces, p<0,01) en células HepG2, mientras que el tratamiento con GW501516 evitó este efecto (p<0,001 vs células estimuladas con IL-6; fig. 1B).
 Extractos de proteína total (STAT3-Tyr705) y nuclear (STAT3-Ser727) se analizaron por Western-blot con anticuerpos contra STAT3 total y fosforilada en Tyr705 y Ser727. Las células se trataron con GW501516 10μmol/l o con vehículo (control) durante 18h antes de ser estimuladas con 20ng/ml de IL-6 durante 10min (STAT3-Ser727) o 2,5h (STAT3-Tyr705). Los valores del gráfico de barras representa el promedio ±DE de 5 experimentos independientes. B) Análisis de los niveles de ARNm de SOCS3 en las células hepáticas humanas tratadas en presencia o en ausencia de GW501516 10μmol/l durante 18h antes del tratamiento de 24h con 20ng/ml de IL-6. Se aisló el ARN total y se analizó por RT-PCR. En la figura se muestra una autorradiografía representativa y la cuantificación normalizada respecto a los niveles de ARNm de 18S. ***p<0,001 vs control; ###p<0,001 vs células estimuladas con IL-6. U.A., unidades arbitrarias; CT, control.")
El agonista de PPARβ/δ, GW501516, reduce los efectos de la IL-6 sobre la fosforilación de STAT3 y la expresión de SOCS3 en células HepG2. A) Extractos de proteína total (STAT3-Tyr705) y nuclear (STAT3-Ser727) se analizaron por Western-blot con anticuerpos contra STAT3 total y fosforilada en Tyr705 y Ser727. Las células se trataron con GW501516 10μmol/l o con vehículo (control) durante 18h antes de ser estimuladas con 20ng/ml de IL-6 durante 10min (STAT3-Ser727) o 2,5h (STAT3-Tyr705). Los valores del gráfico de barras representa el promedio ±DE de 5 experimentos independientes. B) Análisis de los niveles de ARNm de SOCS3 en las células hepáticas humanas tratadas en presencia o en ausencia de GW501516 10μmol/l durante 18h antes del tratamiento de 24h con 20ng/ml de IL-6. Se aisló el ARN total y se analizó por RT-PCR. En la figura se muestra una autorradiografía representativa y la cuantificación normalizada respecto a los niveles de ARNm de 18S. ***p<0,001 vs control; ###p<0,001 vs células estimuladas con IL-6. U.A., unidades arbitrarias; CT, control.
Es bien conocido que la IL-6 activa la ERK1/234, cinasa que fosforila STAT3 en Ser72716. Ya que en nuestro grupo de investigación hemos descrito que el GW501516 evita la activación de ERK1/2 inducida por IL-6 en adipocitos35, evaluamos el efecto de este agonista de PPARβ/δ sobre la activación de esta cinasa en células HepG2. El tratamiento con GW501516 reprimió completamente el aumento de ERK1/2 fosforilada provocado por la presencia de IL-6 en el medio de cultivo (fig. 2A). Utilizamos U0126, un inhibidor específico de ERK1/2 muy potente, para asegurarnos de que la fosforilación de ERK1/2 inducida por IL-6 estaba involucrada en la fosforilación en Ser727 de STAT3. De manera similar a como lo hacía el GW501516, U0126 evitó el aumento en los niveles de fosforilación en Ser727 de STAT3 (fig. 2B). Además, U0126 evitó el aumento en los niveles de ARNm de SOCS3 provocado por IL-6 (fig. 2C), confirmando que la fosforilación de ERK1/2 inducida por IL-6 contribuye a la activación completa de STAT3, llevando a un incremento de la expresión de su gen diana SOCS3.
 o STAT3 total y fosforilada en Ser727 (B). C) Análisis de los niveles de ARNm de SOCS3 en HepG2 en presencia o en ausencia de 10μmol/l GW501516 18h antes de la estimulación con 20ng/ml de IL-6 durante 24h. Se aisló el ARN total y se analizó por RT-PCR. En la figura se muestra una autorradiografía representativa y la cuantificación normalizada respecto a los niveles de ARNm de 18S. Los datos son el promedio ±de de 5 experimentos independientes. **p<0,01 vs control; ***p<0,001 vs control; #p<0,05 vs células estimuladas con IL-6; ##p<0,01 vs células estimuladas con IL-6; ###p<0,001 vs células estimuladas con IL-6. U.A., unidades arbitrarias; CT, control.")
La activación de PPARβ/δ inhibe la fosforilación de ERK1/2 inducida por IL-6. Las células HepG2 fueron tratadas con o sin 10μmol/l de U0126 o GW501516 antes de ser estimuladas con 20ng/ml de IL-6 durante 2,5h. Los lisados celulares se analizaron por Western-blot con anticuerpos para ERK1/2 total y fosforilada (A) o STAT3 total y fosforilada en Ser727 (B). C) Análisis de los niveles de ARNm de SOCS3 en HepG2 en presencia o en ausencia de 10μmol/l GW501516 18h antes de la estimulación con 20ng/ml de IL-6 durante 24h. Se aisló el ARN total y se analizó por RT-PCR. En la figura se muestra una autorradiografía representativa y la cuantificación normalizada respecto a los niveles de ARNm de 18S. Los datos son el promedio ±de de 5 experimentos independientes. **p<0,01 vs control; ***p<0,001 vs control; #p<0,05 vs células estimuladas con IL-6; ##p<0,01 vs células estimuladas con IL-6; ###p<0,001 vs células estimuladas con IL-6. U.A., unidades arbitrarias; CT, control.
Para confirmar el papel de PPARβ/δ en la regulación de la fosforilación de STAT3 utilizamos ratones deficientes para PPARβ/δ. En comparación con los ratones salvajes, los hígados de los ratones deficientes en PPARβ/δ mostraban niveles más elevados de STAT3 fosforilada tanto en Tyr705 como en Ser727 (fig. 3A). Según estos datos, estos ratones mostraron un aumento de ERK1/2 en su estado fosforilado (fig. 3B). Estos resultados demuestran que PPARβ/δ regula tanto ERK1/2 como la fosforilación de STAT3 in vivo.
 (A) y ERK1/2 fosforilada (B). El gráfico de barras muestra los promedios ±DE de 5 experimentos independientes. *p<0,05 vs ratones salvajes; ***p<0,001 vs ratones salvajes. KO, deficientes para Pparβ/δ; WT, «salvajes» (wild type).")
Los ratones deficientes en PPARβ/δ muestran un aumento de la fosforilación de STAT3 y de ERK1/2 en el hígado. Los extractos nucleares de ratones «salvajes» y deficientes en PPARβ/δ se analizaron por Western-blot con anticuerpos contra STAT3 fosforilada (Tyr705, Ser727) (A) y ERK1/2 fosforilada (B). El gráfico de barras muestra los promedios ±DE de 5 experimentos independientes. *p<0,05 vs ratones salvajes; ***p<0,001 vs ratones salvajes. KO, deficientes para Pparβ/δ; WT, «salvajes» (wild type).
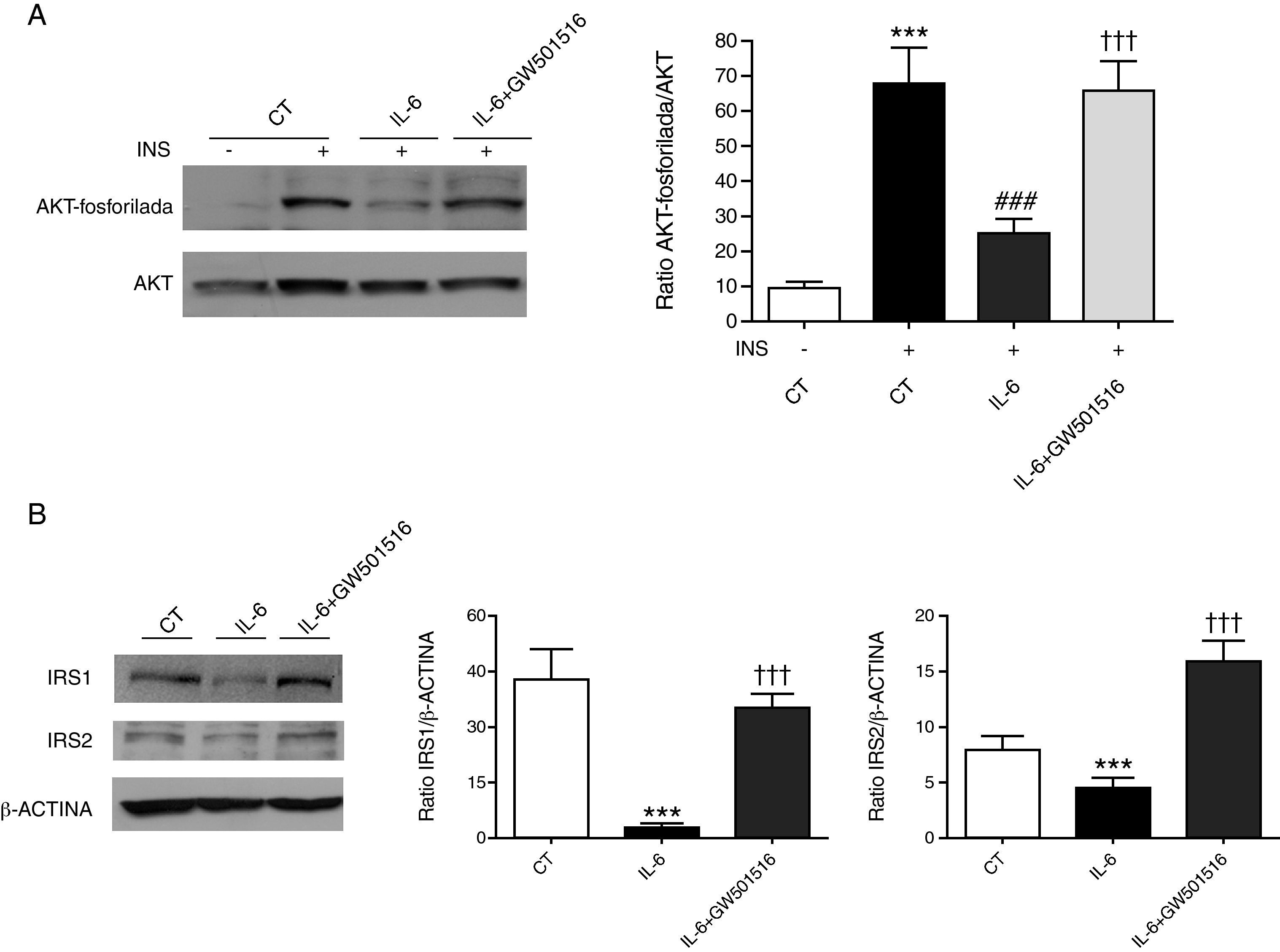
Estudios previos han demostrado que la IL-6 induce RI en células humanas HepG210,11. Para confirmar este efecto estimulamos estas células con esta citocina pro-inflamatoria. Las células expuestas a IL-6 mostraron una reducción significativa de los niveles de AKT fosforilada en presencia de insulina (fig. 4A), confirmando una menor respuesta a esta hormona. Sin embargo, cuando se pre-incubaron las células con IL-6 en presencia de GW501516 se evitó la reducción de la fosforilación de la AKT estimulada por insulina causada por esta citocina. Ya que la RI inducida por IL-6 en células hepáticas se ha atribuido a SOCS310 y esta proteína inhibe la señalización de la insulina a través de la degradación mediada por el proteasoma de IRS-1 e IRS-222, decidimos evaluar sus niveles proteicos. En presencia de IL-6 los niveles proteicos de IRS-1 e IRS-2 eran más bajos que en las células control. Sin embargo, el tratamiento con el agonista de PPARβ/δ evitó estas reducciones (fig. 4B). Estos resultados sugieren que el tratamiento con GW501516 ofrece protección contra los efectos de IL-6 sobre la vía de señalización de la insulina en hepatocitos humanos.
 (A), IRS-1 e IRS-2 (B). Los valores que se muestran en el gráfico son el promedio ±DE de 5 experimentos independientes. *** p<0,001 vs control; ### p<0,001 vs control estimuladas con insulina; ††† p<0,001 vs células estimuladas con IL-6. CT, control; INS, insulina.")
La activación de PPARβ/δ contrarresta la acción de la IL-6 y protege contra los efectos sobre la señalización de la insulina. Las células hepáticas HepG2 se estimularon con insulina 100nmol/l durante 3min, en presencia o ausencia de un pretratamiento con GW501516 10μmol/l durante 18h o IL-6 20ng/ml, o ambos. Los lisados celulares se analizaron por Western-blot utilizando los anticuerpos específicos contra AKT total y fosforilada (Ser473) (A), IRS-1 e IRS-2 (B). Los valores que se muestran en el gráfico son el promedio ±DE de 5 experimentos independientes. *** p<0,001 vs control; ### p<0,001 vs control estimuladas con insulina; ††† p<0,001 vs células estimuladas con IL-6. CT, control; INS, insulina.
La RI y la DM2 están estrechamente asociadas con un estado inflamatorio crónico de baja intensidad caracterizado por la producción anómala de citocinas pro-inflamatorias, como el TNF-α36 y la IL-64,8,37. De estas citocinas, los niveles plasmáticos de IL-6 son los que se correlacionan más fuertemente con la RI5,6. Además, la administración de IL-6 en individuos sanos induce un aumento de la glucosa sanguínea10. Los adipocitos juegan un papel importante en la RI inducida por IL-6, ya que el tejido adiposo es una de las principales fuentes de esta citocina38, la cual puede entrar en el torrente sanguíneo y tener efectos a nivel sistémico modulando la acción de la insulina39. La IL-6 actúa inicialmente activando STAT3 que aumenta la transcripción de su gen diana SOCS3, el cual provoca RI interfiriendo con el receptor de la insulina y con IRS-140. En este estudio hemos demostrado que el agonista de PPARβ/δ, GW501516, puede prevenir la activación de STAT3 inducida por IL-6, así como la consiguiente expresión de SOCS3 en hepatocitos humanos.
La actividad de STAT3 depende de su estado de fosforilación. Así, la fosforilación de STAT3 en el residuo de Tyr705 lleva a la dimerización y posterior translocación al núcleo, donde regula la transcripción de sus genes diana13, mientras que la fosforilación en Ser727 es necesaria para que STAT3 adquiera su máxima actividad transcripcional14,15. Nuestros resultados muestran que el activador de PPARβ/δ GW501516 inhibe la fosforilación de STAT3 inducida por IL-6 en ambos residuos. Esta inhibición parece ser dependiente de PPARβ/δ, ya que en los hígados de ratones deficientes en PPARβ/δ observamos un aumento en los niveles de fosforilación en Tyr705 y Ser727 de STAT3. La prevención de la activación de STAT3 inducida por IL-6 después de la activación de PPARβ/δ puede ser el resultado de diversos mecanismos de acción, como por ejemplo las proteincinasas responsables de la fosforilación en serina de STAT3, entre las que se incluyen, entre otras, la ERK1/236. Previamente nuestro grupo había descrito que GW501516 inhibía la fosforilación de ERK1/2 en adipocitos37. En este estudio hemos observado que GW501516 evita la fosforilación de ERK1/2 inducida por IL-6, lo cual nos sugiere que la inhibición de esta proteincinasa debida a la activación de PPARβ/δ podría ser la responsable de la supresión de la fosforilación en Ser727 de STAT3. En concordancia con el papel de ERK1/2 en la activación de STAT3 hemos demostrado que el uso de U0126, un inhibidor específico de ERK1/2, el cual se une a MEK1/2 (mitogen-activated proteins kinase [MAPK]-ERK1/2) de manera que inhibe su actividad catalítica así como la fosforilación de ERK1/2, reduce la fosforilación en Ser727. Además, U0126 evita el aumento de los niveles de ARNm de SOCS3 inducido por IL-6, sugiriendo que la inhibición de ERK1/2 es suficiente para evitar la activación de la vía STAT3-SOCS3. La reducción en la fosforilación de ERK1/2 provocada por el tratamiento de GW501516 también parece ser dependiente de PPARβ/δ, ya que en los hígados de los ratones deficientes para este receptor se observa un aumento de los niveles de fosforilación de esta cinasa.
De acuerdo con la capacidad de GW501516 para prevenir la expresión de SOCS3 inducida por IL-6 en células HepG2, también hemos demostrado que el tratamiento con este agonista de PPARβ/δ reduce los efectos de la IL-6 sobre la fosforilación de AKT y los niveles proteicos de IRS-1 e IRS-2 inducidos por insulina; de este modo proporcionaría protección contra los efectos de IL-6 sobre la vía de señalización en células hepáticas humanas.
En conjunto, estos resultados sugieren que la inhibición de STAT3 y la consiguiente reducción en los niveles de ARNm de SOCS3 después de la activación de PPARβ/δ en células HepG2 estimuladas con IL-6 podrían contribuir en la prevención de la RI inducida por la IL-6.
Responsabilidades éticasProtección de personas y animales. Los autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datos. Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónEste estudio ha sido financiado parcialmente mediante proyectos de la Swiss National Science Foundation y el Ministerio de Ciencia e Innovación (SAF2009-06939). L. Serrano recibió una beca FPI del Ministerio de Ciencia en Innovación.
AutoríaLucía Serrano-Marco y Manuel Vázquez-Carrera se encargaron del diseño de los experimentos, procesaron las muestras, analizaron y recogieron los datos y redactaron el artículo. Liliane Michalik y Walter Wahli contribuyeron a la interpretación de los datos y revisaron el artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Una comunicación referente a esta línea de trabajo, titulada «El agonista de PPARβ/δ GW501516 evita la activación por interleucina-6 de la vía STAT3-SOCS3 y la reducción de IRS-1 en hepatocitos por inhibición de la ERK1/2», se presentó en el XXIV Congreso Nacional de la SEA (Sevilla 2011) y fue galardonada con una mención especial.