




La aldosterona contribuye al desarrollo de la hipertrofia cardiaca. La quinasa regulada por suero y glucocorticoides 1 (SGK-1) desempeña un papel central en el mecanismo de acción de la aldosterona.
ObjetivoAnalizar las alteraciones cardiacas estructurales, funcionales, inflamatorias y oxidativas, así como la expresión del SGK-1, producidas por la administración de aldosterona+sal en ratas.
Material y métodosSe utilizaron ratas macho Wistar (250g). Un grupo de animales recibió una dosis diaria de aldosterona 1mg/kg/día subcutánea (s.c.)+NaCl al 1% en el agua de bebida. Otro grupo de animales con hiperaldosteronismo inducido fue tratado con un antagonista de los receptores de mineralocorticoides, la espironolactona (200mg/kg/día, s.c.). Como controles se utilizaron animales a los que se les administró el vehículo (s.c.) y animales a los que se les administró espironolactona (200mg/kg/día, s.c.). El periodo de evolución fue de 21días. Se tomaron las siguientes medidas hemodinámicas: PAS, PAD, FC, pDfVI, PSVI, dP/dt+ y dP/dt–. Como índice de hipertrofia cardiaca se calculó el peso relativo del corazón. Se valoró la expresión génica de: TGF-β, CTGF, TNF-α, IL-1α, p22phox, eNOS y SGK-1 y se valoró el contenido de colágeno cardiaco mediante técnicas histológicas.
ResultadosPAS, PAD, pDfVI, PSVI, peso relativo del corazón y el contenido de colágeno aumentaron (p<0,05) en la ratas tratadas con aldosterona+sal. La expresión génica de todos los factores analizados aumentó (p<0,05) con la aldosterona+sal. EL tratamiento con espironolactona normalizó los parámetros modificados por la aldosterona+sal.
ConclusionesEl efecto de la espironolactona disminuyendo el aumento de la expresión incrementada del SGK-1 inducida por la aldosterona indica que la participación de este mediador en las alteraciones cardiacas observadas está relacionada con la activación de los receptores de mineralocorticoides.
Aldosterone induces cardiac hypertrophy and it is known to induce serum and glucocorticoid regulated kinase 1 (SGK-1) gene expression.
AimWe aimed to evaluate structural, functional, inflammatory and oxidative alterations, as well as serum and glucocorticoid regulated kinase1 (SGK-1) expression, produced in rat heart by aldosterone+salt administration. Treatment with spironolactone was evaluated to prove mineralocorticoids mediation.
Matherial and methodsMale Wistar rats received aldosterone (1mg/kg/day)+1% NaCl for 3 weeks. Half of the animals were treated with spironolactone (200mg/kg/day). At the end of treatment hemodynamics were measured: SBP, DBP, LVEDP, LVSP, +dP/dt and –dP/dt. Heart relative weight was measured as cardiac hypertrophy index. mRNA expression of TGF-β, CTGF, MMP2, TIMP2, TNF-α,IL-1β, p22phox, eNOS and SGK-1 were measured. Cardiac collagen content was measured by histological techniques.
ResultsSBP and DBP, LVSP and LVEDP were elevated (P<.05) in aldosterone+salt-treated rats. –dP/dt decreased (P<.05) in aldosterone+salt-treated rats, but +dP/dt was similar in all groups. Spironolactone normalized (P<.05) SBP, DBP, LVSP, LVEDP and –dP/dt. Relative heart weight, collagen content, mRNA expression of TGF-β, CTGF, MMP2, TIMP2, TNF-α, IL-1β, p22phox, eNOS and SGK-1 were increased (P<.05) in aldosterone+salt-treated rats, being reduced by spironolactone (P<.05).
ConlusionsSGK-1 might be a key mediator in the structural, functional and molecular cardiac alterations induced by aldosterone+salt in rats. All the observed changes and mediators are related with activation of mineralocorticoid receptors.
El desarrollo de hipertrofia cardiaca se considera inicialmente una respuesta adaptativa del miocardio al estrés parietal1,2. Los factores que determinan el desarrollo de la hipertrofia cardiaca pueden ser factores hemodinámicos, como la presión arterial, la precarga y la esclerosis de las grandes arterias3,4, o factores no hemodinámicos, como el sistema renina-angiotensina-aldosterona, los factores de crecimiento y las citocinas, entre otros4,5.
La aldosterona contribuye al desarrollo de la hipertrofia cardiaca mediante su efecto hemodinámico. A su vez, se ha demostrado que la aldosterona es uno de los factores mediadores más importantes en el desarrollo de hipertrofia cardiaca, independientemente de los niveles de presión arterial6. La síntesis de colágeno cardiaco está estimulada por la aldosterona y es una de las características principales de la hipertrofia cardiaca producida por el hiperaldosteronismo7,8. Aunque varios estudios se han centrado en el papel del estrés oxidativo y la inflamación en el desarrollo de la hipertorfia cardiaca y la fibrosis producidas por la aldosterona, aún no se conocen todos los mecanismos implicados en su desarrollo. Se ha demostrado que el tratamiento con aldosterona+sal aumenta la expresión de factores profibróticos como el factor de crecimiento transformante (TGF-β) a través mecanismos transcripcionales dependientes del receptor de mineralocorticoides. Además, se han obtenido resultados contradictorios referentes a la participación de otros mediadores de fibrosis cardiaca, como el factor de crecimiento de tejido conectivo (CTGF), las metaloproteasas (MMP) y los inhibidores de metaloproteasas (TIMP) en animales tratados con aldosterona+sal.
La quinasa regulada por suero y glucocorticoides (SGK, serum and glucocorticoid regulated kinase) fue identificada inicialmente como una enzima serina-treonina cinasa inducida de manera rápida por los glucocorticoides en una línea celular de tumor mamario. Se ha comprobado que la aldosterona es capaz de estimular la expresión de SGK-1 a nivel renal de manera precoz (30min) y que esta, a su vez, estimula la actividad del canal epitelial de Na+ (ENaC) y de la Na+/K+ ATPasa en ensayos de coexpresión en oocitos de Xenopus laevis9-12. El estudio del mecanismo de acción de esta cinasa mostró que el SGK-1 desempeña un papel central no solo en el mecanismo de acción de la aldosterona sino también de otros estímulos antinatriuréticos como la insulina13.
Por otra parte, las alteraciones cardiacas producidas por la aldosterona+sal en ratas y su relación con la expresión de SGK-1 incrementada no han sido descritas. El objetivo de nuestro estudio fue analizar las alteraciones cardiacas estructurales, funcionales, inflamatorias y oxidativas, así como la expresión del SGK-1, producidas por la administración de aldosterona+sal en ratas. También se estudió la participación de mediadores profibróticos como el CTGF, MMP-2 y TIMP-2. Como antagonista del receptor de mineralocorticoides se trató a los animales con espironolactona.
Material y métodosDiseño experimental y animalesSe utilizaron 40 ratas macho Wistar (Harlan Ibérica, S.L., Barcelona, España) con un peso inicial medio de 250g. Todos los experimentos se realizaron según las normas de la Universidad Complutense de Madrid y de la Unión Europea para el tratamiento ético de los animales de experimentación. Los animales utilizados en el presente trabajo se mantuvieron en un ciclo de luz-oscuridad de 12h, bajo condiciones controladas de temperatura (20-22°C) y humedad (50-60%) y con acceso libre al agua. Un grupo de animales recibió una dosis diaria de aldosterona de 1.000μg/kg/día. Otro grupo de animales con hiperaldosteronismo fue tratado con un antagonista de los receptores de mineralocorticoides, espironolactona (200mg/kg/día), disuelta en aceite de girasol y mediante una inyección subcutánea. Tanto el grupo de los animales con hiperaldosteronismo como el de los tratados con espironolactona recibieron simultáneamente NaCl al 1% en el agua de bebida. Como controles se utilizaron animales a los que se les administró una inyección subcutánea diaria del vehículo (aceite de girasol) y animales a los que se les administró espironolactona (200mg/kg/día) mediante una inyección diaria. El periodo de evolución fue de 21 días.
Medida de la presión arterial y hemodinámica cardiacaCada animal fue pesado y anestesiado intraperitonealmente con ketamina (Imalgene 1.000) 0,07mg/kg y xilacina (Rompun 2%) 0,024mg/kg. A continuación se introdujo un catéter Sciense FT211B de 1,6F de diámetro (SciSentido, Canadá) por la carótida derecha. Dicho catéter se encontraba acoplado a un sistema de adquisición de datos (PowerLab/800, ADInstruments, Castle Hill, Australia), y este a su vez acoplado a un computador (PC Pentium). Las señales fueron monitorizadas durante 10min a través del programa Chart para Windows v4.2. En primer lugar se registró la presión arterial y a continuación se avanzó el catéter hasta el interior del ventrículo izquierdo, donde se dejó un tiempo de estabilización de 10min. A continuación se comprobó la integridad de la válvula aórtica retirando el catéter del ventrículo y registrando nuevamente la presión arterial. Se tomaron medidas de los siguientes parámetros: presión arterial sistólica (PAS), presión arterial diastólica (PAD), frecuencia cardiaca (FC), presión diastólica final del ventrículo izquierdo (pDfVI), presión sistólica máxima del ventrículo izquierdo (PSVI), derivada positiva de la presión con respecto al tiempo (dP/dt+) y derivada negativa de la presión con respecto al tiempo (dP/dt–). El registro de las señales quedó almacenado para su posterior análisis. Como índice de hipertrofia cardiaca se calculó el peso relativo del corazón: peso corazón/100g peso corporal.
Cuantificación del contenido de colágeno cardiacoSe utilizaron muestras de corazón que se fijaron en etanol al 70% y posteriormente se incluyeron en parafina formando bloques para su tallado en secciones de 4 micras de grosor con un microtomo de rotación (Leitz 1512, IMEB INC, Ch, EE.UU.). A continuación se tiñeron los cortes con rojo picrossirius. El contenido de colágeno se calculó con un analizador de imagen LEICA Q 500IW (Leica, España). Los cortes teñidos se captaron con una videocámara conectada a un microscopio, se digitalizaron y se valoró el colágeno de la muestra.
Análisis por PCR cuantitativa a tiempo realExtracción de ARN mensajeroLos corazones se extrajeron en condiciones estériles y se congelaron inmediatamente en nitrógeno líquido. Para la extracción del ARN mensajero (ARNm) se utilizó un kit de extracción de ARN comercial (Qiagen, Sciences, Maryland, EE.UU.). El ARNm se cuantificó midiendo la densidad óptica a 260nm en un espectrofotómetro (BioPhotometer, Eppendorf, Alemania).
Síntesis de ADN complementarioLa reacción de la transcriptasa reversa se realizó mediante un kit de síntesis de ADNc (Qiagen Sciences, Maryland, EE.UU): 1μg del ARN total se utilizó en la reacción de retrotranscripción. Se eliminó el ADN genómico incubando durante 2min a 42°C el ARN junto con el agua libre de ARNasa y el tampón de lavado del ADN genómico. A continuación se preparó en hielo la mezcla de la transcriptasa reversa, se añadió la muestra de ARN y se incubó durante 15min a 42°C.
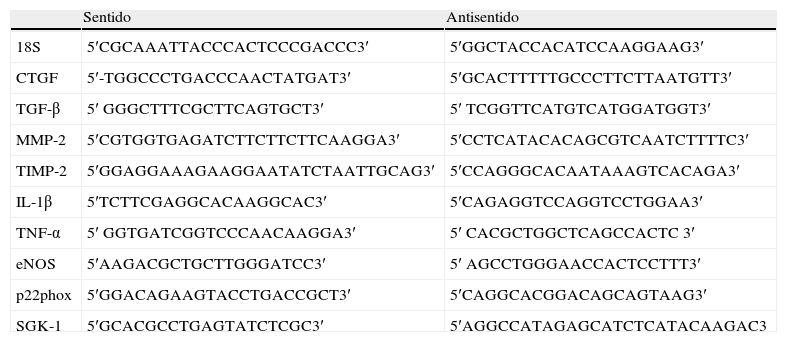
Análisis por PCR cuantitativa a tiempo realPara cuantificar los niveles de ARN mensajero se realizaron reacciones en cadena de la polimerasa cuantitativas y en tiempo real utilizando un termociclador Real Time PCR Smart Cycler (Cepheid, Sunnyvale, California, EE.UU). Para este análisis se utilizaron sondas TaqMan. En las condiciones de anillamiento la sonda hibridó con el ADN molde. En la extensión de las sondas, la actividad exonucleasa de la Taq ADN polimerasa degradó el cebador resultando en la emisión de fluorescencia. La intensidad de fluorescencia correspondió con la cantidad de producto amplificado. Los valores obtenidos se normalizaron con la expresión del 18S utilizado como gen constitutivo y se analizaron mediante el método de 2-ΔΔCT. La tabla 1 recoge las secuencias de los cebadores utilizados.
Secuencias de los cebadores utilizados
| Sentido | Antisentido | |
| 18S | 5′CGCAAATTACCCACTCCCGACCC3′ | 5′GGCTACCACATCCAAGGAAG3′ |
| CTGF | 5′-TGGCCCTGACCCAACTATGAT3′ | 5′GCACTTTTTGCCCTTCTTAATGTT3′ |
| TGF-β | 5′ GGGCTTTCGCTTCAGTGCT3′ | 5′ TCGGTTCATGTCATGGATGGT3′ |
| MMP-2 | 5′CGTGGTGAGATCTTCTTCTTCAAGGA3′ | 5′CCTCATACACAGCGTCAATCTTTTC3′ |
| TIMP-2 | 5′GGAGGAAAGAAGGAATATCTAATTGCAG3′ | 5′CCAGGGCACAATAAAGTCACAGA3′ |
| IL-1β | 5′TCTTCGAGGCACAAGGCAC3′ | 5′CAGAGGTCCAGGTCCTGGAA3′ |
| TNF-α | 5′ GGTGATCGGTCCCAACAAGGA3′ | 5′ CACGCTGGCTCAGCCACTC 3′ |
| eNOS | 5′AAGACGCTGCTTGGGATCC3′ | 5′ AGCCTGGGAACCACTCCTTT3′ |
| p22phox | 5′GGACAGAAGTACCTGACCGCT3′ | 5′CAGGCACGGACAGCAGTAAG3′ |
| SGK-1 | 5′GCACGCCTGAGTATCTCGC3′ | 5′AGGCCATAGAGCATCTCATACAAGAC3 |
Para el análisis estadístico de los resultados se utilizó un test ANOVA de una vía, seguido de un test Newman-Keuls, mediante la utilización de un programa estadístico GraphPad Prism 4. Todos los resultados se consideraron significativos cuando el valor de la p fue menor de 0,05.
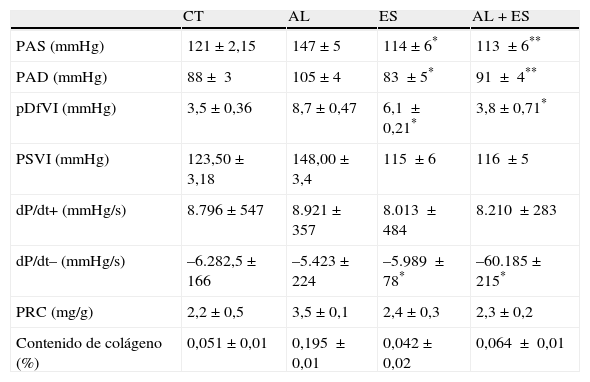
ResultadosParámetros hemodinámicos y morfométricosLa PAS, la PAD y la PSVI aumentaron significativamente en las ratas tratadas con aldosterona+sal comparado con los controles. La pDfVI aumentó y la dP/dt– disminuyó (p<0,05) en las ratas tratadas con aldosterona+sal comparado con los controles, indicando la existencia de disfunción diastólica acompañada por una menor capacidad de relajación del ventrículo izquierdo. El tratamiento con espironolactona redujo (p<0,05) los valores elevados de PAS y PAD y normalizó la pDfVI y la dP/dt–. El tratamiento con espironolactona en ratas a las que no se les administró aldosterona+sal no afectó a ninguno de estos parámetros, excepto a la pDfVI, que fue mayor que la de los controles pero menor que la de las ratas tratadas con aldosterona+sal. La frecuencia cardiaca y la dP/dt– no se modificaron en ninguno de los grupos experimentales estudiados (tabla 2).
Parámetros hemodinámicos y morfométricos
| CT | AL | ES | AL+ES | |
| PAS (mmHg) | 121±2,15 | 147±5 | 114±6* | 113 ±6** |
| PAD (mmHg) | 88± 3 | 105±4 | 83 ±5* | 91 ± 4** |
| pDfVI (mmHg) | 3,5±0,36 | 8,7±0,47 | 6,1 ± 0,21* | 3,8±0,71* |
| PSVI (mmHg) | 123,50± 3,18 | 148,00±3,4 | 115 ±6 | 116 ±5 |
| dP/dt+ (mmHg/s) | 8.796±547 | 8.921±357 | 8.013 ±484 | 8.210 ±283 |
| dP/dt– (mmHg/s) | –6.282,5± 166 | –5.423±224 | –5.989 ±78* | –60.185± 215* |
| PRC (mg/g) | 2,2±0,5 | 3,5±0,1 | 2,4±0,3 | 2,3±0,2 |
| Contenido de colágeno (%) | 0,051±0,01 | 0,195 ±0,01 | 0,042±0,02 | 0,064 ± 0,01 |
AL: aldosterona+sal; AL+ES: aldosterona+sal+espironolactona; CT: control; dP/dt–: derivada negativa de la presión con respecto al tiempo; dP/dt+: derivada positiva de la presión con respecto al tiempo; ES: espironolactona; PAD: presión arterial diastólica; PAS: presión arterial sistólica; pDfVI: presión diastólica final del ventrículo izquierdo; PRC: peso relativo del corazón; PSVI: presión sistólica máxima del ventrículo izquierdo.
*p<0,05 vs. CT; ** p<0,05 vs. ALDO.
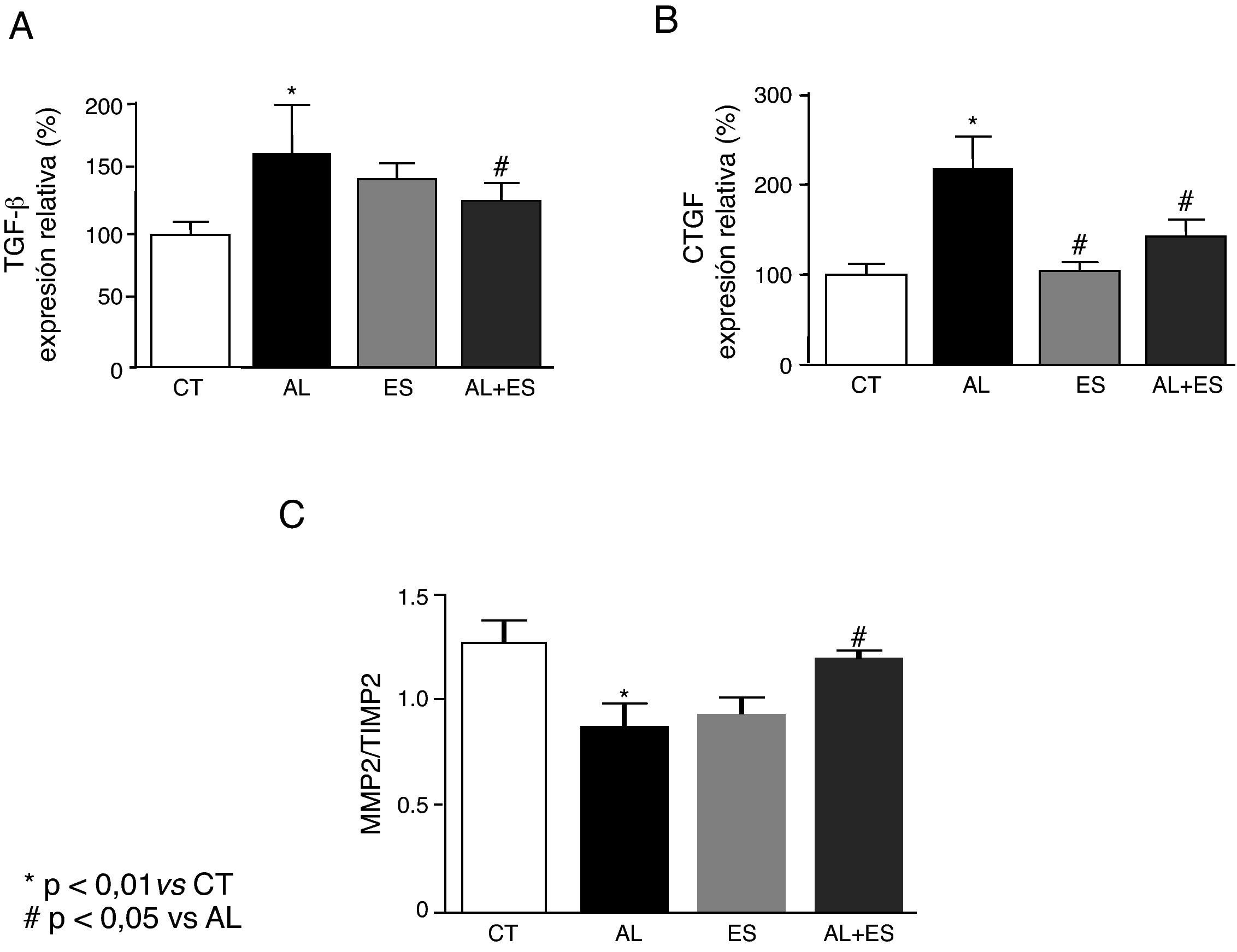
El peso relativo del corazón fue mayor (p<0,05) en las ratas tratadas con aldosterona+sal (tabla 2). Estos animales presentaron también un incremento del contenido de colágeno cardiaco (p<0,05) y un aumento (p<0,05) de la expresión génica de los mediadores fibróticos TGF-β y CTGF comparado con los controles (figs. 1A y B). La expresión génica de MMP-2 y TIMP-2 fue mayor en las ratas tratadas con aldosterona+sal comparado con los controles. La espironolactona redujo (p<0,05) la expresión génica aumentada de todos estos parámetros. La relación MMP-2/TIMP-2 fue menor (p<0,05) en las ratas tratadas con aldosterona+sal comparado con los controles (fig. 1C). El tratamiento con espironolactona aumentó (p<0,05) el valor de esta relación. La espironolactona no afectó a ninguno de estos parámetros en las ratas no tratadas con aldosterona+sal.
 y el CTGF (B) y la relación MMP-2/TIMP-2. AL: aldosterona+sal; AL+ES: aldosterona+sal+espironolactona; CT: control; ES: espironolactona. * p<0,01 vs. CT; # p<0,05 vs. AL.")
Efecto de la administración de aldosterona+sal y tratamiento con espironolactona sobre la expresión génica del TGF-β (A) y el CTGF (B) y la relación MMP-2/TIMP-2. AL: aldosterona+sal; AL+ES: aldosterona+sal+espironolactona; CT: control; ES: espironolactona. * p<0,01 vs. CT; # p<0,05 vs. AL.
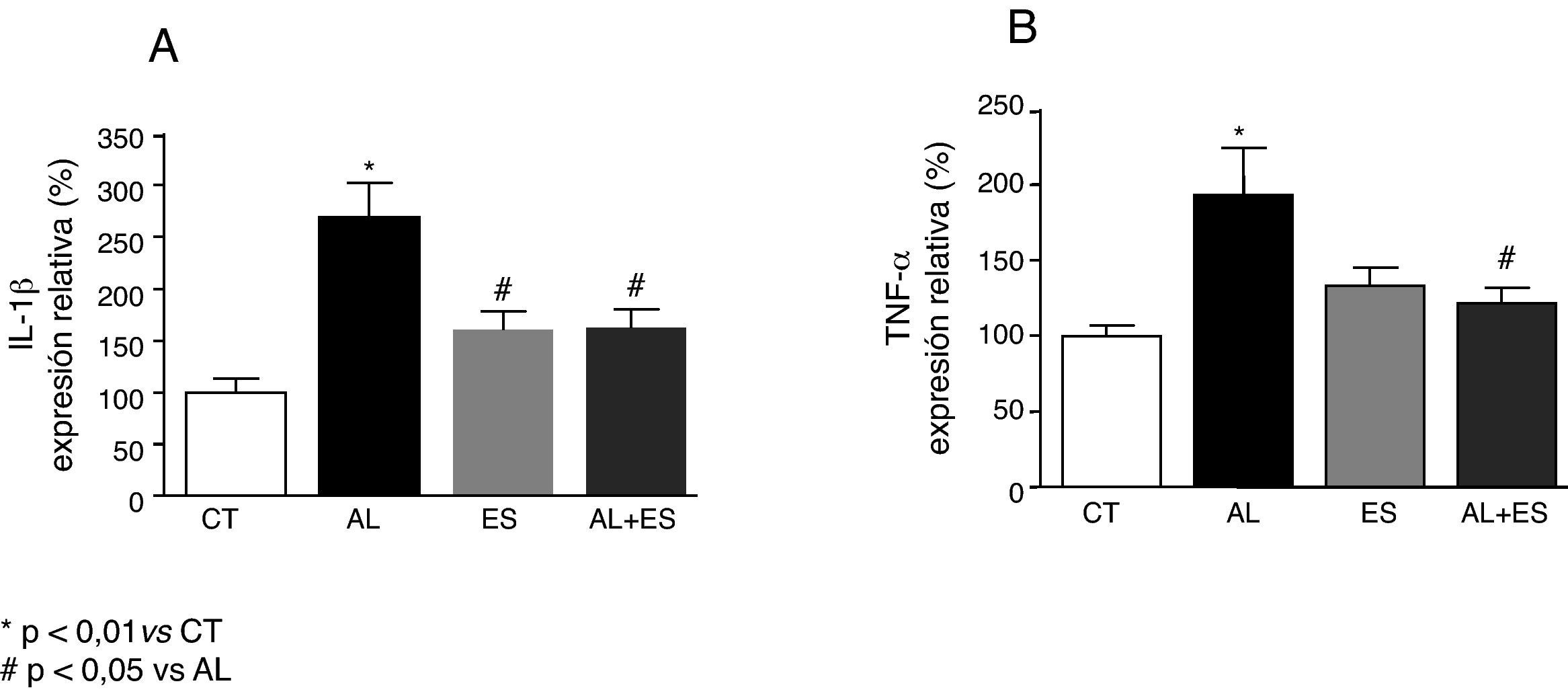
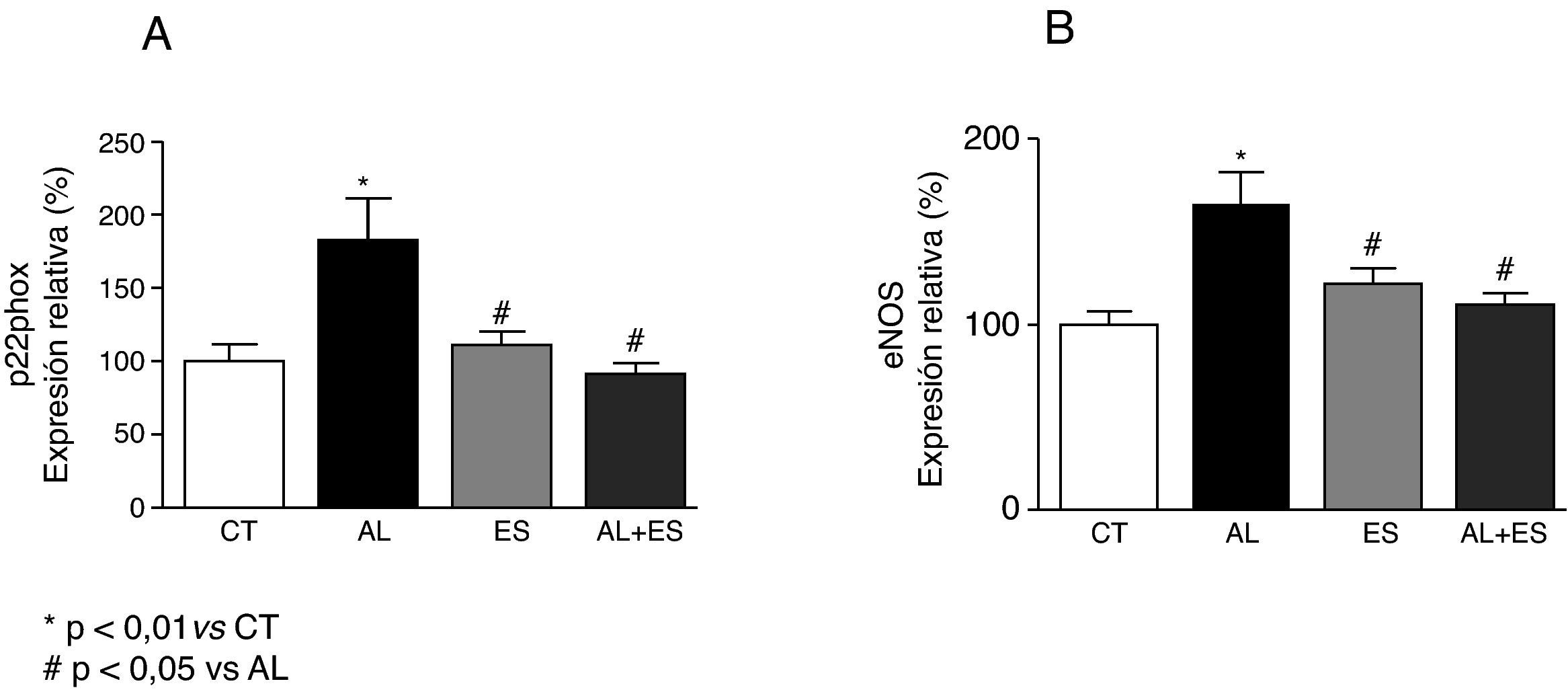
Los niveles de ARNm de TNF-α e IL-1β fueron mayores (p<0,05) en las ratas tratadas con aldosterona+sal que en los controles (figs. 2A y B). De manera similar, las ratas tratadas con aldosterona+sal presentaron niveles de ARNm de p22phox y eNOS incrementados (p<0,05) comparado con los controles (figs. 3A y B). El tratamiento con espironolactona también redujo (p<0,05) estos valores aumentados y no tuvo ningún efecto en las ratas no tratadas con aldosterona+sal.
 y la IL-1β (B). AL: aldosterona+sal; AL+ES: aldosterona+sal+espironolactona; CT: control; ES: espironolactona. * p<0,01 vs. CT; # p<0,05 vs. AL.")
 y la eNOS (B). AL: aldosterona+sal; AL+ES: aldosterona+sal+espironolactona; CT: control; ES: espironolactona. * p<0,01 vs. CT; # p<0,05 vs. AL.")
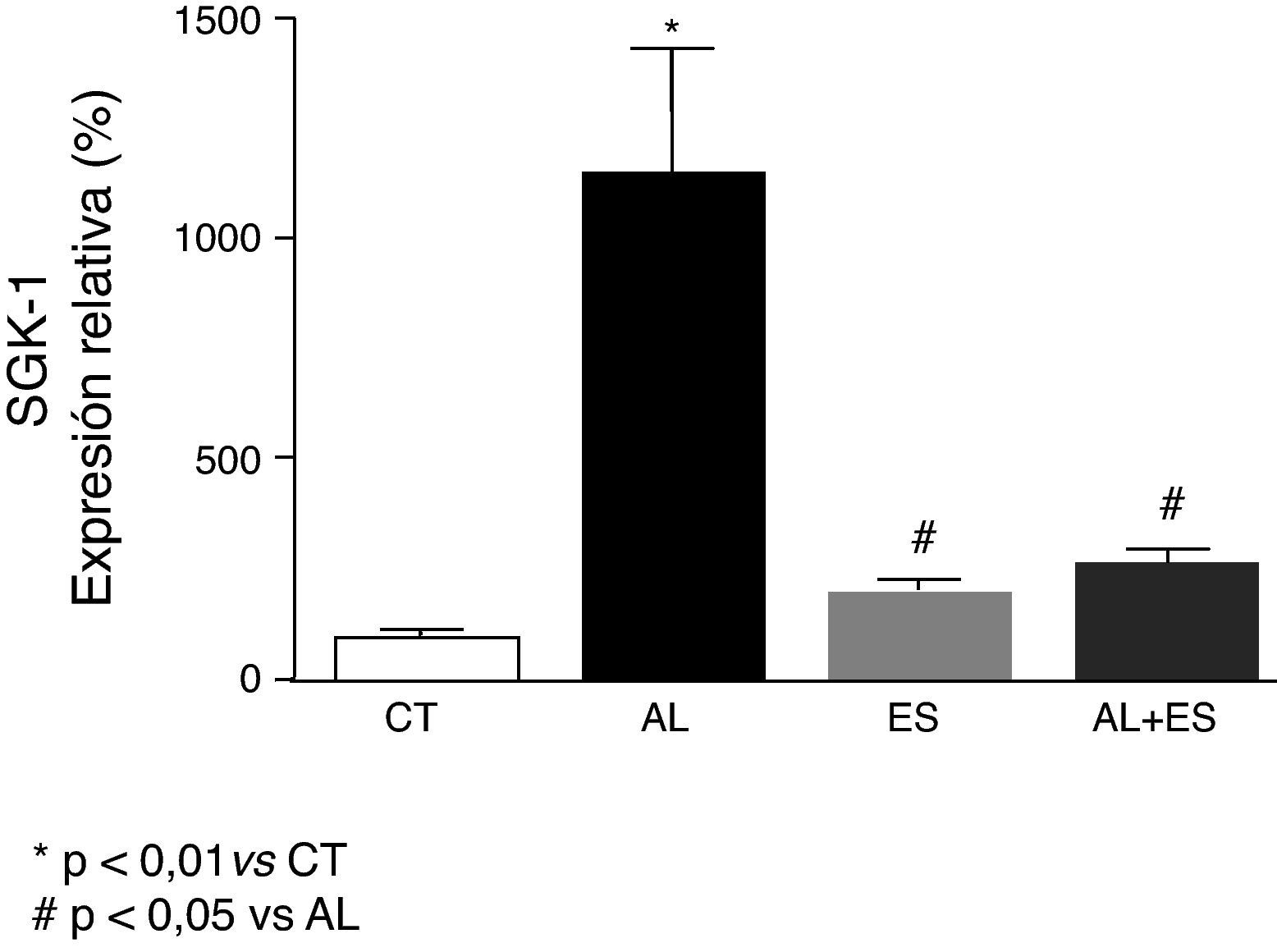
Todos los cambios anteriormente mencionados producidos por la aldosterona+sal estuvieron acompañados por un aumento significativo (p<0,05) de la expresión génica del SGK-1, el cual disminuyó (p<0,05) notablemente con el tratamiento con espironolactona. La espironolactona no modificó la expresión del SGK-1 en las ratas no tratadas con aldosterona+sal (fig. 4).
Discusión
En el presente estudio la administración de aldosterona más 1% de sal a ratas normotensas indujo hipertensión, disfunción diastólica, hipertrofia cardiaca, fibrosis, inflamación, estrés oxidativo y un aumento de la expresión génica del SGK-1 a nivel cardiaco. El tratamiento con el antagonista de los receptores de mineralocorticoides espironolactona bloqueó todos estos efectos.
Las ratas tratadas con aldosterona presentaron niveles aumentados de TNF-α e IL-1β. Numerosas enfermedades cardiacas están asociadas con la expresión de citocinas proinflamatorias como la IL-1β, la IL-6 y el TNF-α, las cuales a través de receptores específicos median en la expresión de determinados genes implicados en el crecimiento celular y en la apoptosis14. En estudios previos se ha demostrado que el tratamiento con aldosterona induce un proceso inflamatorio en las arterias coronarias caracterizado por infiltrado celular de monocitos y macrófagos y por un incremento de la expresión de marcadores inflamatorios como la ciclooxigenasa 2, la osteopontina, la proteína quimiotáctica de macrófagos tipo 1 y la molécula de adhesión intracelular tipo 115. A su vez, la inflamación cardiaca caracterizada en nuestro modelo parece estar relacionada con el aumento del estrés oxidativo cardiaco, como indica el incremento de los niveles del ARNm de la subunidad p22phox de la NADPH oxidasa y de la eNOS que podrían estar induciendo la producción de aniones superóxido.
Numerosos estudios han descrito una relación muy estrecha entre la inflamación y el desarrollo del proceso fibrótico. En el presente estudio, el tratamiento con aldosterona+sal incrementó el peso relativo del corazón y el contenido de colágeno cardiaco relacionados con la disfunción diastólica observada. A través de varios segundos mensajeros (PKC, cAMP, IP3), la aldosterona activa la vía de señalización de la MAPK, la cual promueve la proliferación de fibroblastos16-20. Aunque se ha propuesto que el incremento de colágeno inducido por la aldosterona y secretado por los cardiofibroblastos es independiente de la elevación de la presión arterial, bajo las condiciones experimentales de este estudio no se puede excluir la participación de la hipertensión18. En nuestro estudio encontramos valores aumentados de la expresión génica del mediador de fibrosis TGF-β. Se ha demostrado que el TGF-β también induce fibrosis y remodelado en el corazón, estimulando la transformación de fibroblastos e incrementando la síntesis de las proteínas de matriz extracelular y de integrinas21. En el presente trabajo se demuestra que además del TGF-β, la aldosterona también incrementa la expresión génica del CTGF en corazones de rata. El CTGF ha sido descrito como un factor profibrótico que media algunas de las respuestas del TGF-β, incluidas la apoptosis y la fibrosis22. La sobreexpresión de este factor de crecimiento está asociada a una acumulación de matriz extracelular según se ha descrito en lesiones ateroscleróticas humanas, tras infarto de miocardio y en tejidos vasculares y cardiacos en modelos de hipertensión23-27. En estudios previos se demostró que tanto la aldosterona como el TGF-β inducían preferentemente la expresión del CTGF en cardiomiocitos a través de la activación de las vías ERK1/2, p38 MAPK, y JNK28,29. Se ha demostrado que el CTGF posee efectos pro-hipertróficos en cardiomiocitos al actuar a través de la vía Akt30. En ratas SHR tratadas con eplerenona, la expresión aórtica y renal del CTGF estaba incrementada, sugiriendo la participación de este factor profibrótico en las alteraciones inducidas por la aldosterona asociadas a un estado hipertensivo27,31. El CTGF parece estar mediando en el desarrollo de hipertrofia cardiaca en el presente estudio, a través de la activación del receptor de mineralocorticoides, como sugiere la disminución de estos factores tras el tratamiento con espironolactona.
Así mismo, en el presente trabajo observamos un aumento de la MMP-2. Esta metaloproteasa está expresada ubicuamente en cardiomiocitos y fibroblastos32 y es estimulada también por la IL-1β a través de la vía de la ERK1/2, como se ha sugerido en un estudio previo realizado en cardiofibroblastos33. Nuestros resultados muestran una reducción en la relación MMP-2/TIMP-2 en las ratas tratadas con aldosterona comparado con los controles, sugiriendo una atenuación de la acción de las MMP-2, y consecuentemente una menor degradación de colágeno y un incremento de la fibrosis. Este hecho podría mediar el aumento de colágeno mediante la activación del receptor de mineralocorticoides, como sugiere la disminución de estos factores tras el tratamiento con espironolactona. La participación de las MMP en el remodelado cardiaco a través de los receptores de mineralocorticoides se observó recientemente, en un estudio en el que el tratamiento con espironolactona en ratas SHR con hipertrofia cardiaca disminuía la actividad cardiaca de la MMP-234.
Un mecanismo adicional a los mencionados anteriormente podría ser la expresión aumentada del SGK-1 por el tratamiento con aldosterona+sal y su reducción con el tratamiento con espironolactona. Debido a su efecto dual sobre el incremento de Na+ citosólico y la disminución en la excreción del mismo a nivel renal, cabría esperar un aumento de los niveles de presión arterial35. Por otra parte, se ha demostrado que el SGK-1 induce un incremento de Na+ en el citosol de los cardiomiocitos y los cardiofibroblastos debido principalmente a un incremento en la densidad de sus canales36. Como consecuencia, se producirá un incremento de Ca2+ a través del intercambiador Na+/Ca2+ estimulando la activación de la cascada hipertrófica y los diferentes factores pro-fibróticos, incluidas varias MAPK28,29. Así mismo, el incremento de Na+ citosólico inducido por el SGK-1 puede dar lugar al crecimiento de los cardiomiocitos y por tanto a un incremento del tamaño celular y alteración de sus funciones.
A su vez, el SGK-1 estimula la capacidad del IKKβ para fosforilar al IκBα, dando lugar a la degradación del IκB y por tanto activando al NFκB37. La transcripción de este factor podría estimular el CTGF y la producción de citocinas proinflamatorias a través de las vías explicadas previamente. A la vista de las datos propuestos, podríamos proponer que el SGK-1, a través de la vía del NFκB, estaría incrementado la expresión del CTGF y estimulando el desarrollo de fibrosis cardiaca. Previamente, en un estudio realizado en un modelo DOCA con ratones transgénicos para el SGK-1 y una dieta suplementada con 1% NaCl, se observó un incremento en la síntesis cardiaca de CTGF asociado con un aumento en la expresión del SGK-138. Apoyándonos en esta hipótesis, podríamos proponer también que el SGK-1 podría estar interfiriendo con las MMP y los TIMP, basándonos en el descenso del equilibrio MMP-2/TIMP-2 observado en nuestros resultados, y en el consecuente incremento de colágeno.
Determinados factores, como el TGF-β39-41, el óxido nítrico y las especies reactivas de oxígeno (ERO), también estimulan la expresión del SGK-1. La activación del SGK-1 se produce mediante fosforilación cuando las células se encuentran bajo diferentes estímulos, como puede ser el estrés oxidativo42,43. Como se describió en un estudio previo en el que se analizaba la relación entre el estrés oxidativo y el SGK-1 en un modelo de hipertensión pulmonar, las ERO participan activamente en la regulación de esta cinasa. En dicho estudio se observó que tanto la depleción de la subunidad p22phox de la NADPH oxidasa como el silenciamiento del gen para la proteína Rac inhibían la formación de ERO y prevenían la sobreexpresión del SGK-144. Dado que en el presente estudio hemos observado un incremento en la expresión cardiaca del ARNm del TGF-β, la eNOS, y la NADPH oxidasa, la expresión incrementada del SGK-1 que observamos podría estar a su vez estimulando la expresión de dichos factores (TGF-β, NO y ERO). La reabsorción de Na+ inducida por el SGK-1 estaría incrementando el volumen celular, estimulando así la expresión del TGF-β45.
Por tanto, a la vista de los resultados obtenidos, podemos concluir que el SGK-1 forma parte de un proceso de señalización celular complejo que comprende vías tanto fribróticas como inflamatorias y oxidativas, que estimulan el desarrollo de hipertrofia y fibrosis cardiaca inducida por la aldosterona. Por ello, el SGK-1 parece ser un mediador clave en las alteraciones cardiacas estructurales, funcionales y moleculares inducidas por la aldosterona en ratas. El efecto de la espironolactona disminuyendo el aumento de la expresión incrementada del SGK-1 inducida por la aldosterona indica que la participación de este mediador en las alteraciones cardiacas observadas está relacionada con la activación de los receptores de mineralocorticoides.
Responsabilidades éticasProtección de personas y animales. Los autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datos. Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.