





La hipoxia se considera un factor clave en la progresión de las lesiones ateroscleróticas. El low density lipoprotein receptor-related protein-1 (LRP1) juega un papel fundamental en la vasculatura. El propósito de este estudio fue investigar el efecto de la hipoxia en la expresión y la función del LRP1 en células musculares lisas vasculares (CMLV) y el papel del factor de transcripción inducible por la hipoxia 1 alfa (HIF-1α).
Métodos y resultadosLos análisis por PCR a tiempo real y Western blot demostraron que la hipoxia (1% O2) induce la expresión del LRP1 de manera dependiente del tiempo tanto a nivel de ARN mensajero (niveles máximos al cabo de 1 y 2h) como de proteína (niveles máximos a las 12 y 24h). El retraso en el incremento hipóxico de la proteína del LRP1 en comparación con el ARNm podría explicarse por la larga vida media que presenta la proteína del LRP1. Los ensayos de actividad luciferasa demostraron que la hipoxia y la acumulación de HIF-1α inducen la actividad del promotor del LRP1, y que dos elementos de respuesta a la hipoxia (HRE), situados en las posiciones –1072/–1069 y –695/–692 del promotor, participan en la inducción. La inmunoprecipitación de cromatina mostró la unión in vivo de HIF-1α al promotor del LRP1 en CMLV hipóxicas. Los efectos de la hipoxia en la expresión proteica del LRP1 se tradujeron funcionalmente en un incremento de la acumulación de colesterol esterificado (CE) derivado de la captación de lipoproteínas de baja densidad agregadas (LDLag). El bloqueo de la expresión de HIF-1α inhibió el incremento de la expresión del LRP1 inducido por la hipoxia y la acumulación de CE intracelular derivado de las LDLag, sugiriendo que tanto la sobreexpresión del LRP1 como la sobreacumulación de CE en células vasculares hipóxicas dependen de HIF-1α. Los análisis inmunohistoquímicos mostraron que el LRP1 y el HIF-1α colocalizan en células vasculares de placas ateroscleróticas avanzadas.
ConclusionesLa hipoxia incrementa la expresión del LRP1 y la acumulación intracelular de CE derivado de LDLag en CMLV humanas a través de la acumulación de HIF-1α.
Hypoxia is considered a key factor in the progression of atherosclerotic lesions. Low density lipoprotein receptor-related protein 1 (LRP1) plays a pivotal role in the vasculature. The aims of this study were to investigate the effect of hypoxia on LRP1 expression and function in vascular smooth muscle cells (VSMC) and the role of hypoxia-inducible factor alpha (HIF-1α).
Methods and resultsReal time PCR and Western blot analysis demonstrated that hypoxia (1% O2) time-dependently induced LRP1 mRNA (maximum levels at 1-2hours) and protein expression (maximum levels at 12-24hours). The delayed hypoxic upregulation of LRP1 protein versus mRNA may be explained by the long half-life of LRP1 protein. Luciferase assays demonstrated that hypoxia and HIF-1α over-accumulation induced LRP1 promoter activity and that two consensus HRE sites located at –1072/–1069 and –695/–692 participate in the induction. Chromatin immunoprecipitation showed the in vivo binding of HIF-1α to LRP1 promoter in hypoxic VSMC. Hypoxia effects on LRP1 protein expression were functionally translated into an increased cholesteryl ester (CE) accumulation from aggregated LDL (agLDL) uptake. The blockade of HIF-1α expression inhibited the upregulatory effect of hypoxia on LRP1 expression and agLDL-derived intracellular CE over-accumulation, suggesting that both LRP1 overexpression and CE over-accumulation in hypoxic vascular cells are dependent on HIF-1α. Immunohistochemical analysis showed the colocalization of LRP1 and HIF-1α in vascular cells of human advanced atherosclerotic plaques.
ConclusionsHypoxia upregulates LRP1 expression and agLDL-derived intracellular CE accumulation in human VSMC through HIF-1α induction.
La hipoxia juega un papel fundamental en la fisiopatología del cáncer, del infarto de miocardio y de la aterosclerosis. El engrosamiento de la pared arterial asociado a la progresión de la placa aterosclerótica reduce el aporte de oxígeno en ciertas áreas de la íntima vascular1,2. La hipoxia está presente en lesiones ateroscleróticas avanzadas humanas y se correlaciona con la presencia de angiogénesis y de trombosis3,4. La mayoría de las células en zonas hipóxicas de placas ateroscleróticas responden alterando la expresión de genes involucrados en vasculogénesis, angiogénesis, inflamación y deposición lipídica5-11. Estos genes se activan a través del factor de transcripción inducible por la hipoxia 1 (HIF-1). HIF-1 es un complejo que se forma por la unión de las subnidades HIF-1α y HIF-1β. La subunidad HIF-1α está sujeta a una rápida degradación proteosomal dependiente de la presencia de oxígeno mediante la hidroxilación de residuos de prolina por hidrolasas12-14. En presencia de bajas concentraciones de oxígeno, HIF-1α se acumula y dimeriza con la subunidad HIF-1β, la cual se expresa de forma constitutiva, permitiendo que el complejo HIF-1 se una a las secuencias HRE (elementos de repuesta a la hipoxia) de los promotores de genes diana15. Se ha descrito que la acumulación de HIF-1α en macrófagos promueve la formación de células espumosas y el desarrollo de la aterosclerosis16. En modelos experimentales de hipercolesterolemia e hipertensión, HIF-1α también está asociado a células musculares lisas vasculares (CMLV)17,18. La hipoxia modula procesos clave para la funcionalidad de las CMLV, como la proliferación/migración 19,20 y la acumulación lipídica21. La agregación y la fusión de las lipoproteínas de baja densidad (LDLag), uno de los principales inductores de la formación de células espumosas derivadas de CMLV22-24, se sabe que incrementa en condiciones de hipoxia.25 El low density lipoprotein receptor-related protein 1 (LRP1), receptor lipoproteico capaz de unir e internalizar LDLag23,24, incrementa por hipercolesterolemia26 e hipertensión27. Las LDLag estimulan la transcripción del LRP1 disminuyendo los niveles de los sterol regulatory element binding protein (SREBP)26. El SREBP-1 y el SREBP-2 son factores de transcripción que modulan negativamente la transcripción del LRP1 en CMLV humanas28,29. El incremento del LRP1 por factores de riesgo cardiovascular podría contribuir a la sobreexpresión del LRP1 que se encuentra en las lesiones ateroscleróticas avanzadas30-32. Se desconoce si la hipoxia puede modular la expresión del LRP1 en las placas de ateroma humanas, y si es así, no se sabe qué factores podrían estar implicados.
Los objetivos de este trabajo fueron investigar el efecto de la hipoxia en la expresión y la función del LRP1 vascular y los mecanismos moleculares implicados en este efecto.
Material y métodosCultivos celularesLos cultivos de CMLV humanas se obtuvieron a partir de la capa media de arterias coronarias macroscópicamente sanas recogidos de pacientes sometidos a trasplantes cardiacos en el Hospital de la Santa Creu i Sant Pau de Barcelona. Las CMLV fueron aisladas con una modificación de la técnica de explantes previamente descrita22-24. Los explantes se incubaron a 37°C en una atmósfera húmeda con un 5% de CO2. Después de una semana, las células empezaron a migrar de los explantes y a proliferar cubriendo la superficie de la placa de cultivo. El medio se renovó cada 2 días una vez comenzó el crecimiento de las células, y al cabo de 10 días se obtuvo una alto grado de proliferación. Los explantes se recogieron con pinzas y se colocaron en placas nuevas con medio de cultivo fresco hasta que las células alcanzaron un alto grado de confluencia. Para la caracterización celular se utilizaron marcadores específicos de diferenciación tales como α-actina (45 kDa) y calponina (33 kDa). Las monocapas celulares crecieron en medio 199 suplementado con un 20% de suero fetal bovino (FBS), un 2% de suero humano, 2mmol/l de L-glutamina, 100U/ml de penicilina G y 100μg/ml de estreptomicina. Las células quiescentes (expuestas durante 24h en medio de cultivo con solo un 0,2% de FBS) se sometieron a normoxia (21% O2) en un incubador Nirco con una mezcla de gases del 74% N2 y un 5% CO2, o a hipoxia (1% O2) en una estación de hipoxia Hypoxic/Anoxic Workstation H35 (Don Whitley Scientific Ltd.) con una mezcla de gases de un 1% O2, 94% N2 y 5% CO2. El estudio fue aprobado por el comité ético institucional del Hospital de la Santa Creu i Sant Pau y llevado a cabo de acuerdo con la Declaración de Helsinki.
Las células HeLa se mantuvieron en medio DMEM con 100U/ml de penicilina G y 100μg/ml de estreptomicina, suplementado con un 10% de FBS.
Silenciamiento génico del LRP1 y HIF-1α mediante ARN de interferenciaPara inhibir la expresión del LRP1 en CMLV humanas, las células se transfectaron transitoriamente con un ARN pequeño de interferencia (siARN-LRP1) sintetizado por Ambion de acuerdo con unas secuencias diana del LRP1 previamente publicadas por nuestro grupo23,24. El siARN-LRP1 se trataba de un oligonucleótido de 15 pares de bases (pb), 5’-CGGCGGGGTCAGCAT-3’, complementario a los nucleótidos comprendidos entre las posiciones 466 y 481 del ARN mensajero (ARNm) del LRP1. Se utilizó un siARN-aleatorio como control negativo (Ambion, AM 4636) en las transfecciones celulares. El análisis Fasta en las bases de datos génicas mediante el Genetic Computer Group Package indicó que estas secuencias no deberían hibridar con otras secuencias del LRP1 ni con secuencias de otros receptores de las LDL. Para el silenciamiento de HIF-1α se utilizó un siARN específico (siARN-HIF-1α) de Applied Biosystems (siARN ID 42840). Las CMLV quiescentes se transfectaron con los siARN mediante la técnica de nucleofección utilizando el kit Human AoSMC Nucleofector de Amaxa de acuerdo con las instrucciones del fabricante. La concentración final de los siARN para la transfección fue de 0,6μmol/l. Al cabo de 48h después de la transfección, las células se expusieron a normoxia o hipoxia durante los tiempos de estudio y se recogieron mediante raspado en el reactivo TriPure (Roche) para análisis de PCR y Western blot.
Aislamiento y modificación de LDLLas LDL humanas (LDL sin modificar o LDL nativas con densidad entre d1,019-d1,063g/ml) se obtuvieron a partir de una mezcla de sueros de donantes normocolesterolémicos mediante ultracentrifugaciones secuenciales. Las LDL se dializaron y se determinó la concentración proteica utilizando el método del ácido bicinconínico. La concentración de colesterol se determinó con un kit comercial (Boehringer). Las LDL se utilizaron en los experimentos antes de que pasasen 48h desde su aislamiento. La pureza de las LDL se analizó con un gel de electroforesis (Paragon System, Beckmann). Las LDLag se obtuvieron por agitación intensa de las LDL nativas (LDLn) diluidas en tampón fosfato salino (PBS). La formación de los agregados de LDL se monitorizaron por turbidimetría (absorbancia a 680nm), como ya se había descrito previamente33. La ultraestructura de las LDLag obtenidas por agitación intensa fue similar a las LDL modificadas por versicán24, uno de los principales proteoglicanos estructurales condroitín sulfato de la capa íntima arterial.
Determinación del contenido intracelular de colesterol libre, colesterol esterificado y triglicéridosLas CMLV quiescentes cultivadas en condiciones de normoxia o hipoxia durante 24h se expusieron durante las últimas 12h a concentraciones crecientes de LDL. Después del periodo de incubación con las lipoproteínas, las células se lavaron exhaustivamente: 2 veces con PBS, 2 veces con PBS-1% albúmina de suero bovino (BSA) y 2 veces con PBS-1%BSA-100 U/ml heparina antes de ser recogidas en 1ml de NaOH 0,15mol/l. La extracción lipídica se realizó utilizando el método de Bligh y Dyer34 con algunas modificaciones menores.33 Las muestras y concentraciones diferentes de estándares (una mezcla de colesterol, triglicéridos y colesterol palmitato) se aplicaron en placas de cromatografía en capa fina. Las placas se secaron y los puntos se tiñeron de acuerdo con Huber et al.35. Los puntos de la placa cromatográfica correspondientes al colesterol libre (FC), a los triglicéridos (TG) y a los ésteres de colesterol (CE) se cuantificaron por densitometría mediante la utilización de la curva de estándares y de un densitómetro (Molecular Dynamics).
Mutagénesis dirigida en construcciones del promotor LRP1Para este estudio se construyó un plásmido con el promotor del LRP1 unido al gen reportero de la luciferasa. Inicialmente se amplificó por PCR un fragmento del promotor del gen del LRP1 que contenía la secuencia que va desde la posición –1305 hasta +341 (considerando que la transcripción empieza en el nucleótido +1) con los cebadores: 5¿-CTGTTTCTGCCAAGGACAGACTGG-3’ y 5’-CTGGTGCGCTTTGCCGAAGGAAAGA-3’. El fragmento se clonó en el vector pGL3 (Promega) entre las zonas de restricción NheI-HindIII. El ADN para la PCR se obtuvo de células humanas sanguíneas. La secuencia del promotor comentada anteriormente (GI: 34338) y que contiene los lugares de unión para determinados factores de transcripción se analizó mediante el programa Matinspector (Genomatix). Este programa mostró dos posibles secuencias HRE (HRE-1: desde –1072 hasta –1069, y HRE-2: desde –695 hasta –692) dentro del promotor del LRP1.
Para evaluar la relevancia de estas posibles secuencias HRE en la modulación del LRP1 por la hipoxia, realizamos una serie de deleciones en la construcción del promotor del LRP1 con el fin de eliminar la posible HRE-2 o las dos posibles HRE. Las deleciones se realizaron con los siguientes cebadores directos: 5’-ACCCGCTGGTGACTCACCTCCTCCAA-3’ (que elimina la HRE-2) o 5’-CCTAGAGTATGACACTGAGTTT-3’ (que elimina las dos secuencias HRE), y sus correspondientes cebadores reversos complementarios. Para mutar la secuencia HRE-1 en la posición –1072 y la HRE-2 en la posición –695 seleccionamos los siguientes cebadores directos:
5’-GGCTCCATCCCCGAGCCCTTTTCGGGCGGACAAGCTCC-3’ y 5’-GGGGCAGTGACCAAAAGTTTTTTCACTGGCCCCTGGGAACCG-3’ (las bases mutadas aparecen subrayadas), y sus correspondientes oligonucleótidos reversos complementarios. Se utilizó el kit de mutagénesis dirigida Quickchange II (Stratagene). Los plásmidos resultantes se aislaron y sus secuencias se confirmaron por digestión con enzimas de restricción y secuenciación del ADN.
Transfecciones transitorias para estudios de actividad del promotor LRP1Para estudiar la actividad transcripcional del promotor humano del LRP1 en condiciones de hipoxia, se transfectaron cultivos de células HeLa con las construcciones truncadas, mutadas y wildtype descritas anteriormente, y se sometieron a normoxia o hipoxia. Para comprobar que el efecto de la hipoxia se debía a la acción de HIF-1α, se realizaron experimentos de cotransfección en los que se transfectó, además de las construcciones con el promotor, un vector de sobreexpresión de HIF-1α (pHIF-1α, cedido por el Dr. Luis del Peso, Departamento de Bioquímica de la Universidad Autónoma de Madrid) en condiciones de normoxia o un vector vacío (pcADN3). En las transfecciones se utilizó el reactivo Lipofectamina-Plus (Invitrogen).
En los ensayos de cotransfección, las células se recogieron en tampón de lisis y la sobreexpresión de HIF-1α se analizó por Western blot. Las transfecciones se realizaron en placas de 12 pocillos con 0,5μg de ADN plasmídico/pocillo y 2μl de lipofectamina durante 3h. Transcurridas las 3h, se cambió el medio y se incubó durante 24h. Al día siguiente las células se llevaron a quiescencia durante 24h más y finalmente se expusieron a normoxia o hipoxia durante 18h. Los extractos celulares se recogieron según las indicaciones del kit Dual Luciferase assay (Promega). La lectura se realizó utilizando un luminómetro de placas Berthold-Orion. Los valores de la actividad luciferasa se normalizaron respecto a la concentración total de proteína. En las transfecciones en las que se analizó la actividad basal realizamos cotransfecciones con un plásmido que tiene clonado el gen de la Renilla. En estos experimentos los valores de luciferasa se normalizaron por la actividad Renilla.
Determinación de la estabilidad del ARNm y de la proteína del LRP1 en condiciones de normoxia e hipoxiaPara determinar si la hipoxia podría alterar la estabilidad del ARNm del LRP1, las CMLV quiescentes se expusieron durante 4h a normoxia o hipoxia. Después se añadieron a los cultivos 4,5μg/ml de actinomicina D (un inhibidor de la transcripción celular) y se devolvieron a las concentraciones de O2 en las que se encontraban. Las células se recogieron en TriPure (Roche) a diferentes tiempos (1, 2, 3, 6 y 12h) después de la adición de actinomicina D. Después de purificar el ARN total, la estabilidad del ARNm del LRP1 se determinó por PCR a tiempo real y se calculó como el porcentaje del ARNm inicial que quedaba después de la exposición a actinomicina D.
Para determinar si la sobreexpresión del LRP1 por la hipoxia dependía de la actividad transcripcional, las CMLV controles y pretratadas con actinomicina D durante 4h se expusieron a normoxia o hipoxia durante 16h y se recogieron en tampón de lisis (10μM Tris-HCl pH7,5; 150μM KCl; 5%TritonX-100) suplementado con el cóctel de inhibidores de proteasas Complete (Roche) para estudios de Western blot.
Finalmente, para estudiar la estabilidad de la proteína del LRP1 y determinar su vida media, los cultivos de CMLV quiescentes se expusieron durante 18h a normoxia o hipoxia. Después se añadieron 100μmol/l de cicloheximida (un inhibidor de la síntesis proteica) a los cultivos y se expusieron de nuevo a las concentraciones de O2 en las que se encontraban. Las células se recogieron en tampón de lisis a diferentes tiempos (1, 2, 3, 6, 12 y 32h) después de la adición de cicloheximida para la realización de Western blot. La estabilidad de la proteína del LRP1 se calculó como la proporción de la proteína inicial restante después del tratamiento con cicloheximida.
Estudios de expresión génica por PCR a tiempo realPara la extracción del ARN total, las células se lavaron con PBS y se recogieron en el reactivo TriPure (Roche) siguiendo las indicaciones del fabricante. Este método de aislamiento se basa en la diferencia de solubilidad del ARN, del ADN y de la proteína en distintos solventes orgánicos. La integridad del ARN total se comprobó mediante electroforesis en gel de agarosa al 1% con bromuro de etidio 100μg/ml. La cuantificación del ARN total se realizó leyendo la absorbancia de las muestras a la longitud de onda de 260nm en un espectrofotómetro NanoDrop ND-1000 (NanoDrop Technologies).
La síntesis del ADN de cadena sencilla copia (cADN) del ARNm presente en la célula se realizó a partir de ∼1,5μg de ARN total por retrotranscripción utilizando el kit High Capacity cADN Reverse Transcription (Applied Biosystems). En este protocolo se utilizaron secuencias aleatorias como cebadores y la enzima MultiscribeTM (incluida en el kit) como transcriptasa reversa.
El análisis de la expresión génica se llevó a cabo por PCR a tiempo real en el equipo PCR-700026 Sequence Detection System de ABIPRISM (Applied Biosystems).
La cuantificación de la expresión génica por PCR se realizó utilizando la TaqMan Universal Master Mix (Applied Biosystems) y una mezcla comercial de sonda y cebadores (Assays on Demand, Applied Biosystems) específica para cada gen de estudio. Los assays utilizados se detallan a continuación: LRP1 (Hs00233899_m1), receptor clásico de las LDL o LDLR (Hs00181192_m1), SREBP-1 (Hs00231674_m1) y SREBP-2 (Hs00190237-m1). El gen TBP (4326317E), factor de transcripción de unión a cajas TATA del ADN, se usó como control endógeno.
Análisis proteico por Western blotPara la extracción de la proteína total las células se lavaron con PBS y se recogieron en el reactivo (Roche) TriPure siguiendo las indicaciones del fabricante. Se obtuvo la proteína siguiendo el método de aislamiento del TriPure según las indicaciones del fabricante, al finalizar se resuspendió en dodecilsulfato sódico (SDS) al 1% y se cuantificó mediante el ensayo del ácido bicinconínico. Las proteínas se separaron en un gel de poliacrilamida al 10% desnaturalizante (con SDS) y se transfirieron a una membrana de nitrocelulosa. La membrana se bloqueó y se incubó con anticuerpos monoclonales contra el LRP1 humano (cadena β del LRP1; Research Diagnostics, clon 8B8 RDI 61067, dilución 1:40) o contra HIF-1α humano (BD Biosciences, 610958, dilución 1:500). El control de la proteína total cargada en la electroforesis se realizó mediante la tinción de Ponceau de las membranas y la detección del gen endógeno de la β-actina (Abcam, ab8226-100, dilución 1:5000).
InmunocitoquímicasLas CMLV se sembraron en cubres de vidrio, se cultivaron y se sometieron a normoxia o hipoxia durante 18h. Las células fijadas se permeabilizaron y se incubaron con un anticuerpo primario de ratón anti-LRP1 humano (cadena-β, Research Diagnostics, clon 8B8, RDI 61067, dilución 1:5). Las placas se lavaron y se incubaron con el anticuerpo secundario de cabra anti-IgG de ratón conjugado al fluoróforo FITC (dilución 1:100). Las imágenes de las células marcadas se analizaron a través de un microscopio confocal invertido de fluorescencia (Leica TCS SP2-AOBS, Wetzlar) y se procesaron con el programa informático Leica Standard Software TCS-AOBS. La superficie fluorescente se calculó usando el programa NIH Image software (dominio público, desarrollado por el Dr. Wayne Rasband, National Institutes of Health, versión 1.6, pixel2/célula).
InmunohistoquímicasLas arterias coronarias obtenidas de los corazones explantados se cortaron en secciones seriadas de 5μm de grosor. Los anticuerpos primarios utilizados fueron monoclonales de ratón: anti-LRP1 (PRO61067; Fitzgerald), anti-HIF-1α (Abcam; ab8366) y anti-α-actina (Dako; M851). Después de la incubación con los anticuerpos, se lavaron las secciones, se suprimió la actividad peroxidasa con H2O2 y se bloquearon las uniones inespecíficas con suero equino. Los anticuerpos primarios se detectaron usando la técnica de la inmunoperoxidasa gracias a la afinidad avidina-biotina. Las secciones se incubaron con los anticuerpos secundarios biotinilados apropiados (Vector®). El cromógeno usado fue 3,3’-diaminobenzidine. Se utilizó hematoxilina para las tinciones nucleares.
Para los estudios de colocalización se aplicó un método de inmunofluorescencia indirecta. Las secciones se incubaron con anticuerpos primarios monoclonales de ratón anti-HIF-1α (Abcam, ab8366) y el monoclonal de conejo anti-LRP1 (Epitomics, 2703-1) durante 2h a temperatura ambiente. Después de varios lavados con PBS (100mM)/ Triton X-100 al 0,1%, las secciones se incubaron con FITC (515-095-003; Jackson) y con TRITC (R156; Dako) durante 1h a temperatura ambiente. Entonces se lavaron con PBS 100mmol/l, pH7,4 y se montaron con medio Glicergel® (Dako A/S). Las imágenes se capturaron en el microscopio a mediante la cámara Retiga 1300i Fast.
Ensayos de inmunoprecipitación de cromatinaPara la realización de la inmunoprecipitación de cromatina (ChIP) se siguieron las indicaciones del kit ExactaChip de R&D Systems (ECP19335). Las CMLV quiescentes y sometidas a normoxia o hipoxia se fijaron durante 15min a temperatura ambiente con formaldehído al 37% a una concentración final del 1%. La reacción de fijación se detuvo incubando con glicina 5min a temperatura ambiente a una concentración final de 125mM. A partir de aquí, las muestras se mantuvieron siempre en hielo y con una mezcla de inhibidores de proteasas (10μg/ml leupeptina, 10μg/ml aprotinina y 1mM phenylmethanesulfonyl fluoride [PMSF]; todos de Roche). Se lavaron las células tres veces con PBS e inhibidores de proteasas, se recogieron en PBS y, después de centrifugar, se lisaron con tampón de lisis (suministrado por el kit) más inhibidores de proteasas durante 10min.
Los lisados celulares se sonicaron para fragmentar el ADN en el baño del sonicador Bioruptor UCD-200 de Diagenode, el cual está asociado a un sistema de refrigeración de agua Neslab RTE7 de Thermo Scientific. Se usaron 4 ciclos de sonicación de 15min. A través de un gel de agarosa se comprobó que los fragmentos de ADN obtenidos tras la sonicación tenían un tamaño que iba desde los 800 a los 200pb. Para esto se cargó un 0,5% de cada muestra sonicada en el gel.
Una vez fragmentado el ADN, se centrifugaron las muestras durante 10min a 4°C, se descartó el pellet con los restos celulares y se continuó el ChIP con los sobrenadantes, que es donde se encuentra el ADN. Se tomó una muestra de cada sobrenadante (un 10% del volumen) como input o ADN total de partida con el que se normalizaron los resultados finales. Los sobrenadantes se diluyeron con tampón de dilución (suministrado por el kit) y se incubaron un mínimo de 2h a 4°C en un agitador orbital con 5μg del anticuerpo específico anti-HIF-1α o IgG de cabra (control negativo), ambos anticuerpos suministrados por el kit y biotinilados.
Los fragmentos de ADN unidos a HIF-1α reconocidos por el anticuerpo anti-HIF-1α se recuperaron añadiendo bolas magnéticas (MAG999 de R&D con estreptavidina). Después de sucesivos lavados con tampones suministrados por el kit, se añadió una solución de resina quelante (suministrada en el kit) y se hirvieron las muestras durante 10min para liberar el ADN inmunoprecipitado de las bolas y de los anticuerpos. Finalmente, se precipitaron las bolas unidas a los anticuerpos y se recuperaron los sobrenadantes con el ADN.
El ADN se purificó mediante una extracción con fenol-cloroformo y realizó una PCR a tiempo real con SYBR green de Roche en un equipo PCR-7900 Sequence Detection System de ABIPRISM (Applied Biosystems) para las secuencias HRE. Se usaron los siguientes cebadores:
Para la secuencia HRE-1 (–726/–529) el cebador directo 5¿-GGTACTAAGGTGGGCTCCAT-3’ y el reverso: 5¿-AGGCTGAGCTTTGATTTG GA-3’. Para la secuencia HRE-2 (–1192/–1035) el directo: 5’-GCTCCTTGAACTCTGACATGC-3’ y el reverso: 5’-CCCACACGGGAATGATACTT-3’. Se amplificó durante 40ciclos de 95°C 15s y 60°C 1min. Como control positivo, se realizó una PCR a tiempo real para el VEGF humano con cebadores suministrados por el kit.
Los threshold cycle (Ct) de todas las muestras fueron normalizados en función de los inputs de partida (ΔCt). Finalmente, los resultados de las muestras inmunoprecipitadas con el anticuerpo anti-HIF-1α se corrigieron por los resultados de las IgG (ΔΔCt).
Análisis estadísticosLos resultados se expresaron como la media±SEM y el número de experimentos se mostró en cada caso. Las diferencias estadísticas entre los controles y los grupos tratados se analizaron por el test no paramétrico de Mann-Whitney para datos pareados. Se consideró significante un valor de p igual o menor a 0,05.
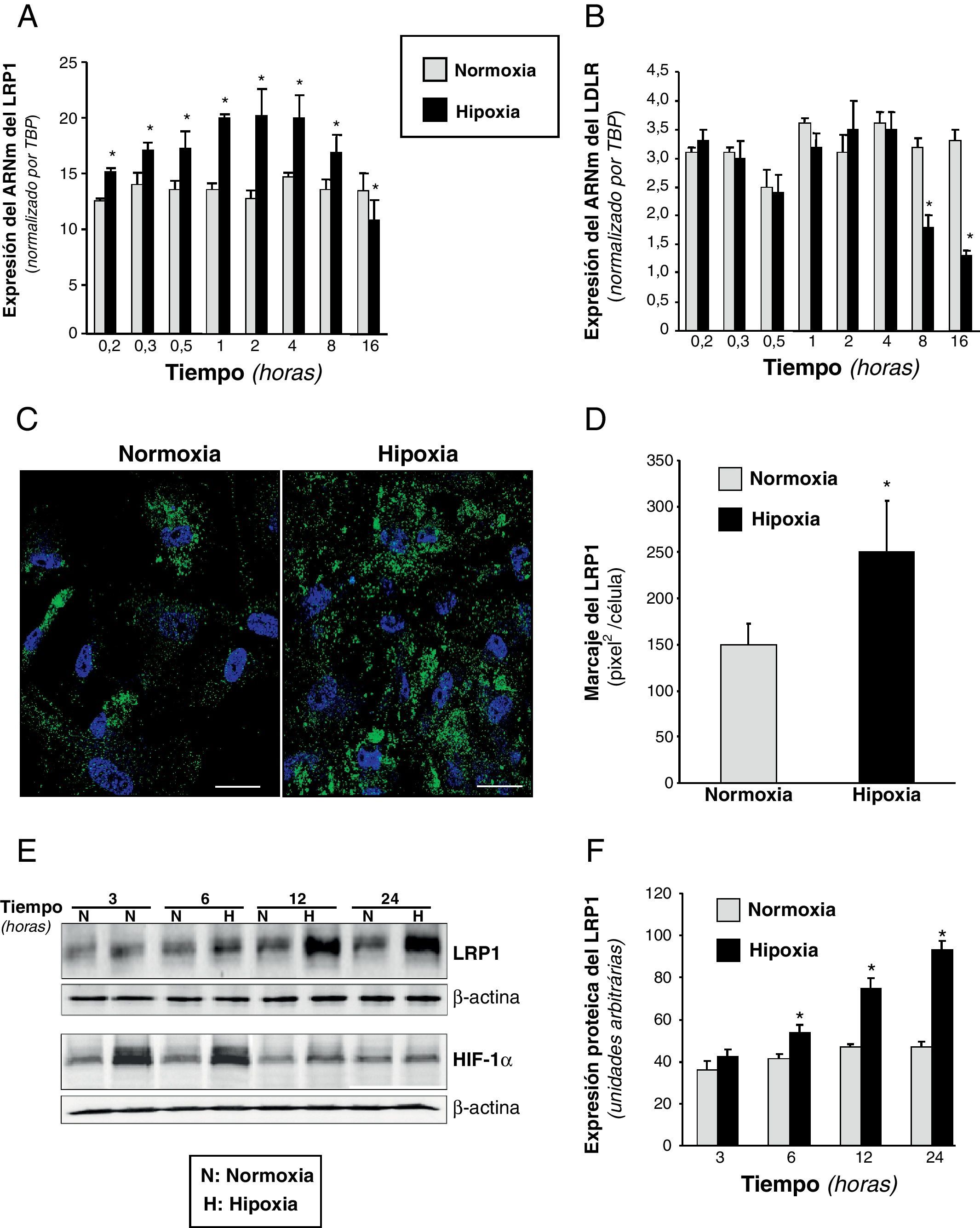
ResultadosLa hipoxia incrementa la expresión del LRP1 en CMLV humanasLos resultados de PCR a tiempo real (fig. 1A) demostraron que la hipoxia inducía la expresión del ARNm del LRP1 en CMLV de una forma dependiente del tiempo. La máxima expresión del ARNm del LRP1 (1,8 veces aproximadamente) se alcanzó después de 60min de hipoxia y decayó después de 16h. El ARNm del LDLR se indujo levemente al cabo de 1h y decayó hasta niveles basales a partir de las 8h de hipoxia (fig. 1B). Los niveles de expresión del ARNm del SREBP-1 y del SREBP-2 fueron significativamente disminuidos, un 26±3% y un 54±2,5%, respectivamente, después de 16h de hipoxia (tabla 1). Experimentos de microscopia confocal demostraron que los niveles del LRP1 eran más elevados en el citoplasma de CMLV hipóxicas que en las células normóxicas (fig. 1C). La cuantificación de la superficie cubierta por la fluorescencia mostró que las CMLV hipóxicas tenían aproximadamente 1,7 veces más marcaje que las células normóxicas (hipoxia: 250±56 vs normoxia: 150±22, pixel2/célula) (fig. 1D). Análisis por Western blot demostraron que la hipoxia incrementa los niveles proteicos del LRP1 y de HIF-1α, aunque a diferentes tiempos (fig. 1E). Los niveles de la proteína del LRP1 se incrementaron desde 1,35 a las 6hs hasta 2 a las 24h de hipoxia (fig. 1E-F). Por el contrario, los niveles proteicos de HIF1-α alcanzaron el máximo a tiempos cortos (fig. 1E). Estos resultados muestran diferencias a lo largo del tiempo en el incremento celular del LRP1 y del HIF-1α en respuesta a situación de hipoxia.
 y del receptor de las LDL (LDLR) (B). Los datos se procesaron con un programa informático específico que utiliza los valores de los Ct y se normalizaron por el ARNm del gen endógeno (TBP). Los resultados están expresados como la media±SEM de 3 experimentos independientes realizados por duplicado (* p<0,05 vs CMLV en normoxia). C) Las microfotografías de microscopia confocal muestran el marcaje del LRP1 en CMLV cultivadas en condiciones de normoxia o hipoxia durante 18h. Las células se lavaron, se fijaron, se permeabilizaron y se incubaron con anticuerpos anti-LRP1 (escala de barra: 10μm). D) El diagrama de barras muestra la cuantificación de la superficie cubierta por la fluorescencia en las CMLV normóxicas e hipóxicas (pixel2/célula). Los resultados se muestran como la media±SEM de 5 imágenes pertenecientes a 4 experimentos independientes. E) Western blot del LRP1 y de HIF-1α en CMLV expuestas a normoxia o hipoxia a lo largo del tiempo. Los niveles de α-actina se muestran como control de carga. F) Diagrama de barras de la cuantificación de las bandas del LRP1 en CMLV normóxicas e hipóxicas. Los resultados se expresan como la media±SEM de 3 experimentos independientes realizados en duplicado (* p<0,05 vs células control).")
Efecto de la hipoxia en la expresión del ARNm y de la proteína del LRP1 en CMLV humanas. Se expusieron CMLV a normoxia o hipoxia durante tiempos crecientes. Se muestran los niveles de ARNm obtenidos por PCR a tiempo real del LRP1 (A) y del receptor de las LDL (LDLR) (B). Los datos se procesaron con un programa informático específico que utiliza los valores de los Ct y se normalizaron por el ARNm del gen endógeno (TBP). Los resultados están expresados como la media±SEM de 3 experimentos independientes realizados por duplicado (* p<0,05 vs CMLV en normoxia). C) Las microfotografías de microscopia confocal muestran el marcaje del LRP1 en CMLV cultivadas en condiciones de normoxia o hipoxia durante 18h. Las células se lavaron, se fijaron, se permeabilizaron y se incubaron con anticuerpos anti-LRP1 (escala de barra: 10μm). D) El diagrama de barras muestra la cuantificación de la superficie cubierta por la fluorescencia en las CMLV normóxicas e hipóxicas (pixel2/célula). Los resultados se muestran como la media±SEM de 5 imágenes pertenecientes a 4 experimentos independientes. E) Western blot del LRP1 y de HIF-1α en CMLV expuestas a normoxia o hipoxia a lo largo del tiempo. Los niveles de α-actina se muestran como control de carga. F) Diagrama de barras de la cuantificación de las bandas del LRP1 en CMLV normóxicas e hipóxicas. Los resultados se expresan como la media±SEM de 3 experimentos independientes realizados en duplicado (* p<0,05 vs células control).
Efecto de la hipoxia sobre los niveles de expresión del ARNm del SREBP-1 y SREBP-2 en células musculares lisas vasculares (CMLV) humanas
| Tiempo de exposición a 1%O2 (horas) | SREBP-1 ARNm | SREBP-2 ARNm |
| 0 | 1,35±0,20 | 63,10±4,12 |
| 4 | 1,31±0,13 | 65,22±1,42 |
| 8 | 1,34±0,16 | 51,00±3,43* |
| 16 | 1,02±0,31* | 29,02±1,9* |
CMLVs quiescentes se expusieron a normoxia (21% O2) o hipoxia (1% O2) a diferentes tiempos. Se cuantificaron los niveles de expresión del ARNm del SREBP-1 y del SREBP-2 por PCR a tiempo real a cada tiempo de estudio. Los datos de la PCR se procesaron con un programa informático específico que utiliza los valores de los Ct y los normaliza por el ARNm del gen endógeno (TBP). Los resultados se expresaron como la media±SEM de 3 experimentos realizados por duplicado.
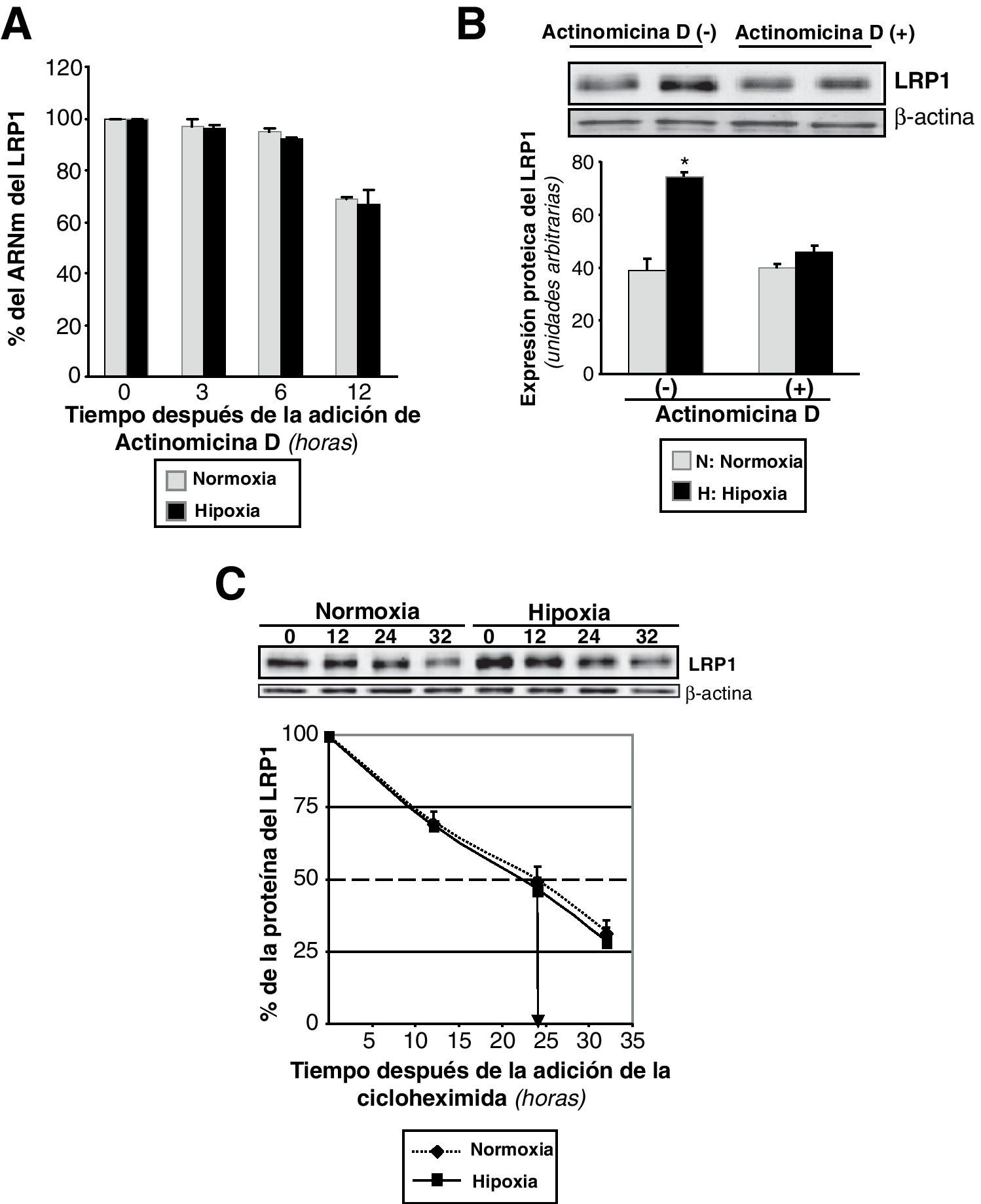
Para comprobar si la hipoxia podría estar influyendo la estabilidad del ARNm del LRP1, se trataron las CMLV con actinomicina D, un inhibidor de la transcripción celular. Inicialmente se preexpusieron las células quiescentes a normoxia o a hipoxia durante 4h. Entonces se añadió actinomicina D (4,5μg/ml) y se devolvieron las células a normoxia o hipoxia. Las células se recogieron a diferentes tiempos después del tratamiento y se procesaron para analizar los niveles de expresión del ARNm del LRP1. La expresión del ARNm permaneció constante durante las 6 primeras horas de tratamiento (datos no mostrados). Al cabo de 12h desde la adición de la actinomicina D, el ARNm del LRP1 decayó fuertemente tanto en normoxia como en hipoxia (normoxia: hasta un 69%±0,5; hipoxia: hasta un 67%±5) (fig. 2A). Para confirmar que el incremento de la proteína del LRP1 por la hipoxia dependía de la transcripción, CMLV sin tratar y tratadas con actinomicina D se expusieron a hipoxia. Como se muestra en la figura 2B, el incremento hipóxico del LRP1 se previno completamente en presencia de actinomicina D.
 CMLV quiescentes se preexpusieron a normoxia o hipoxia durante 4h. Después se añadió actinomicina D (4,5μg/ml) y se continuó la exposición de las células a normoxia e hipoxia, respectivamente. Las células fueron recogidas a los tiempos indicados y el ARNm fue analizado por PCR a tiempo real. El valor de expresión del LRP1 a las 0h se estableció como el 100% y se utilizó para determinar el porcentaje de ARNm del LRP1 restante después de la adición de la actinomicina D. Los resultados están expresados como la media±SEM de 2 experimentos independientes realizados por triplicado. B) CMLV se incubaron en presencia o ausencia de actinomicina D (4,5μg/ml) y se expusieron a normoxia o hipoxia durante 16h. Las células se recogieron y se procesaron para Western blot, como se explica en la metodología. Los resultados están expresados como la media±SEM de 2 experimentos independiente realizados por triplicado (*p<0,05 vs células en normoxia). C) CMLV se preexpusieron a normoxia o hipoxia durante 24h y seguidamente se incubaron con cicloheximida (100μmol/l) durante los tiempos indicados. Las células se recogieron y procesaron para Western blot. Los niveles de α-actina se muestran como control de carga. El gráfico de líneas muestra el porcentaje de proteína del LRP1 restante después de la adición de la cicloheximida. Los resultados están expresados como la media±SEM de 2 experimentos independientes realizados por triplicado. Las flechas indican el tiempo en que la proteína se encuentra reducida al 50% (vida media).")
Efecto de la hipoxia en la estabilidad del ARNm y de la proteína del LRP1. A) CMLV quiescentes se preexpusieron a normoxia o hipoxia durante 4h. Después se añadió actinomicina D (4,5μg/ml) y se continuó la exposición de las células a normoxia e hipoxia, respectivamente. Las células fueron recogidas a los tiempos indicados y el ARNm fue analizado por PCR a tiempo real. El valor de expresión del LRP1 a las 0h se estableció como el 100% y se utilizó para determinar el porcentaje de ARNm del LRP1 restante después de la adición de la actinomicina D. Los resultados están expresados como la media±SEM de 2 experimentos independientes realizados por triplicado. B) CMLV se incubaron en presencia o ausencia de actinomicina D (4,5μg/ml) y se expusieron a normoxia o hipoxia durante 16h. Las células se recogieron y se procesaron para Western blot, como se explica en la metodología. Los resultados están expresados como la media±SEM de 2 experimentos independiente realizados por triplicado (*p<0,05 vs células en normoxia). C) CMLV se preexpusieron a normoxia o hipoxia durante 24h y seguidamente se incubaron con cicloheximida (100μmol/l) durante los tiempos indicados. Las células se recogieron y procesaron para Western blot. Los niveles de α-actina se muestran como control de carga. El gráfico de líneas muestra el porcentaje de proteína del LRP1 restante después de la adición de la cicloheximida. Los resultados están expresados como la media±SEM de 2 experimentos independientes realizados por triplicado. Las flechas indican el tiempo en que la proteína se encuentra reducida al 50% (vida media).
Para medir la estabilidad proteica, se preexpusieron CMLV humanas a normoxia o hipoxia y seguidamente se les adicionó cicloheximida para bloquear la traducción. La expresión proteica del LRP1 permaneció constante durante las 12 primeras horas de tratamiento con cicloheximida (datos no mostrados). Sin embargo, el porcentaje inicial de la proteína del LRP1 decayó fuertemente a partir de las 12h (normoxia: hasta un 70%±3,42; hipoxia: hasta un 69%±4,67) y hasta las 32h (normoxia: hasta un 32%±3,76; hipoxia: hasta un 29%±4,45) (fig. 2C).
Estos resultados sugieren que la vida media de la proteína del LRP1 es de 24h, independientemente de que las condiciones sean normóxicas o hipóxicas. De forma adicional, estos resultados muestran que tanto la actividad transcripcional como la translacional son necesarias para el incremento del LRP1 por hipoxia.
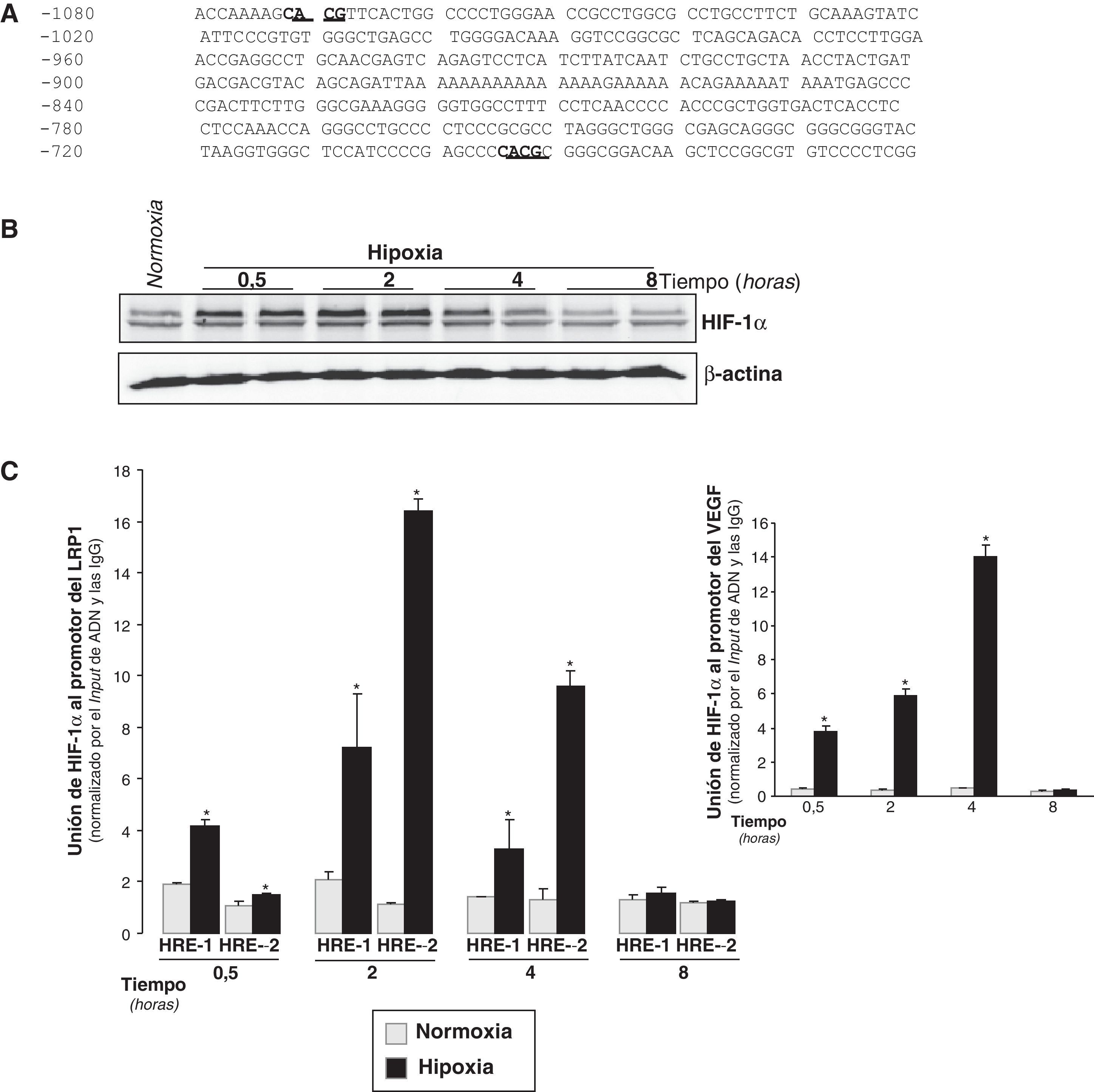
La hipoxia incrementa los niveles de HIF-1α y su unión al promotor LRP1 en CMLV humanasSe analizó el promotor del gen del LRP1 mediante la utilización del programa MatInspector (Genomatix) y se detectó la presencia de dos posibles secuencias HRE (CACG): HRE-1 y HRE-2 en las posiciones –695/–692 y –1072/–1069 respectivamente (fig. 3A).
 Representación esquemática de la región del promotor del LRP1 con 2 posibles secuencias HRE en las posiciones –1072/–1069 (HRE-2) y –695/–692 (HRE-1), indicadas con el texto subrayado. B) Representación de un Western blot que muestra los niveles proteicos de HIF-1α con el tiempo de exposición de CMLV a hipoxia. Los niveles de α-actina se muestran como controles de carga. C) El ensayo de inmunoprecipitación de cromatina muestra la asociación relativa de HIF-1α a las secuencias HRE-1 y HRE-2 del promotor del LRP1 in vivo. La cromatina se rompió por sonicación y las inmunoprecipitaciones se realizaron con un anticuerpo anti-HIF-1α o con IgG no específicas de cabra como control negativo. La unión relativa de HIF-1α in vivo se cuantificó por PCR a tiempo real utilizando cebadores específicos para el promotor del LRP1. También se realizaron PCR de los inputs (ADN total de partida). Los Ct de las muestras inmunoprecipitadas con el anticuerpo específico anti-HIF-1α y con las IgG no específicas se normalizaron por los Ct de los inputs (ΔCt). Los resultados obtenidos con el anticuerpo específico se normalizaron por aquellos obtenidos con las IgG (ΔΔCt). Los resultados están expresados como la media±SEM de 3 experimentos realizados en duplicado (*p<0,01 vs controles en normoxia).")
Inducción de HIF-1α y de su unión al promotor del LRP1 con el tiempo de exposición de CMLV humanas a hipoxia. CMLV quiescentes se expusieron tanto a normoxia como a hipoxia durante 0,5, 2, 4 y 8h. A) Representación esquemática de la región del promotor del LRP1 con 2 posibles secuencias HRE en las posiciones –1072/–1069 (HRE-2) y –695/–692 (HRE-1), indicadas con el texto subrayado. B) Representación de un Western blot que muestra los niveles proteicos de HIF-1α con el tiempo de exposición de CMLV a hipoxia. Los niveles de α-actina se muestran como controles de carga. C) El ensayo de inmunoprecipitación de cromatina muestra la asociación relativa de HIF-1α a las secuencias HRE-1 y HRE-2 del promotor del LRP1 in vivo. La cromatina se rompió por sonicación y las inmunoprecipitaciones se realizaron con un anticuerpo anti-HIF-1α o con IgG no específicas de cabra como control negativo. La unión relativa de HIF-1α in vivo se cuantificó por PCR a tiempo real utilizando cebadores específicos para el promotor del LRP1. También se realizaron PCR de los inputs (ADN total de partida). Los Ct de las muestras inmunoprecipitadas con el anticuerpo específico anti-HIF-1α y con las IgG no específicas se normalizaron por los Ct de los inputs (ΔCt). Los resultados obtenidos con el anticuerpo específico se normalizaron por aquellos obtenidos con las IgG (ΔΔCt). Los resultados están expresados como la media±SEM de 3 experimentos realizados en duplicado (*p<0,01 vs controles en normoxia).
Los resultados de Western blot mostraron que los niveles proteicos de HIF-1α de CMLV estaban significantemente inducidos a los 30min de exposición a hipoxia, y que alcanzaban el máximo después de 2h (fig. 3B).
Los ensayos de inmunoprecipitación de cromatina realizados en CMLV demostraron que HIF-1α se une a las secuencias HRE-1 y HRE-2 en el promotor del LRP1 in vivo (fig. 3C). Aunque la unión de HIF-1α a las dos secuencias ya se detectaba a los 30min de hipoxia, la máxima unión a ambos elementos se alcanzó transcurridas las 2h de hipoxia. La unión de HIF-1α a la secuencia HRE-2 fue superior que a la HRE-1 a las 2 y a las 4h. Como era de esperar36,37, encontramos una unión muy fuerte de HIF-1α al promotor de VEGF (control positivo) después de 30min de hipoxia (fig. 3C).
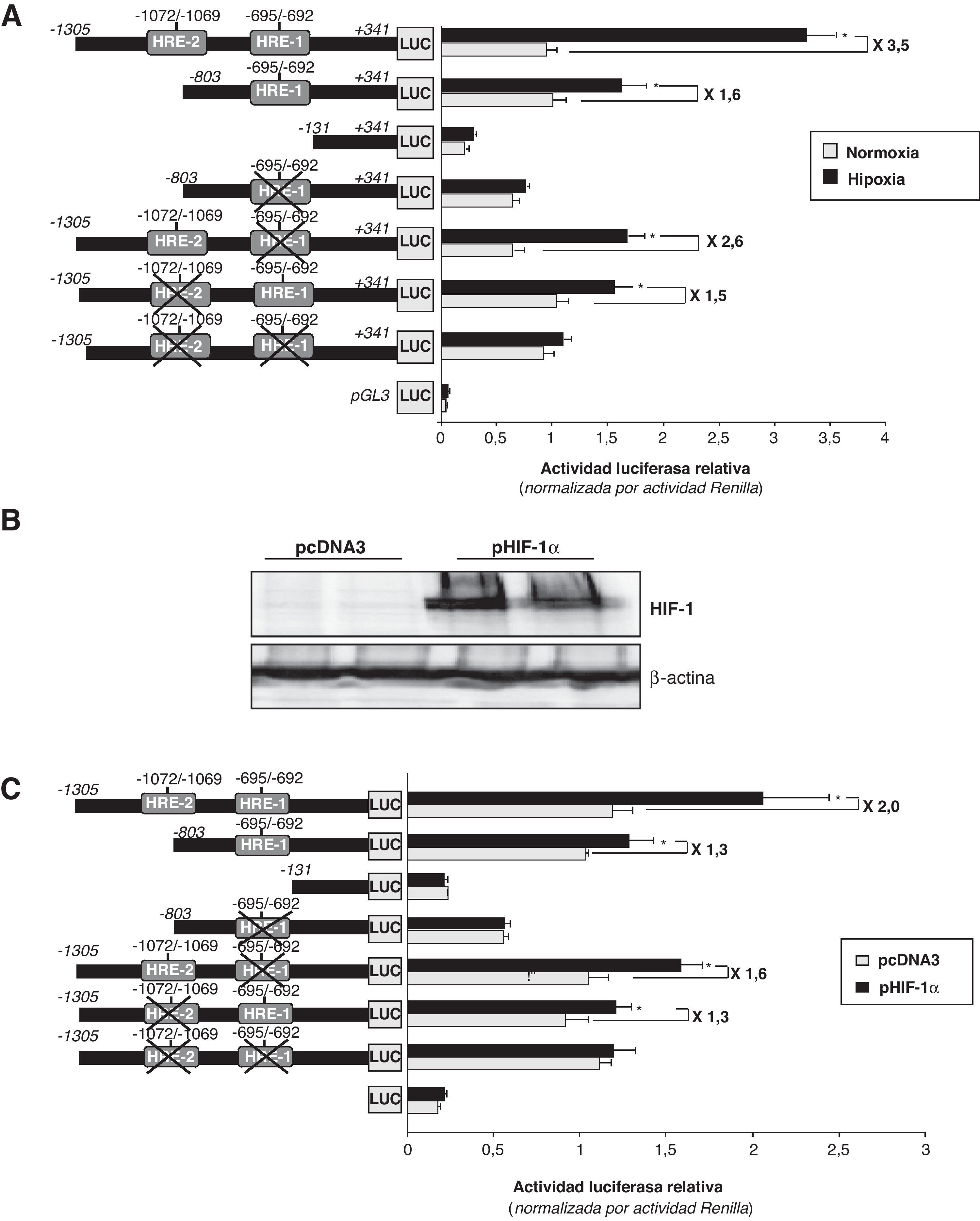
La hipoxia incrementa la actividad transcripcional del promotor LRP1 a través de dos secuencias HREPara investigar el papel de las secuencias HRE-1 y HRE-2 en la modulación hipóxica del promotor del LRP1, realizamos transfecciones transitorias utilizando construcciones del promotor del LRP1 con las dos secuencias HRE (de –1305 a +341), con la HRE-1 (de –803 a +341), sin secuencias HRE (de –131 a +341) y con las dos secuencias HRE mutadas. Utilizamos células HeLa debido a que son un modelo ideal para realizar transfecciones38,39 y analizar los lugares de unión del complejo HIF1 en el promotor de genes que se inducen por la hipoxia. Como se muestra en la figura 4A, la hipoxia (24h) incrementó 3,5 veces la actividad transcripcional del promotor salvaje del LRP1, y 2,6 veces la del promotor con la HRE-1 mutada. La hipoxia, sin embargo, solo incrementó moderadamente la actividad del promotor mutado en la HRE-2 (menos de 2 veces) y no ejerció ningún efecto en la construcción sin secuencias HRE.
 Diferentes regiones del promotor del LRP1 (de –1035 a +341, de –183 a +341, y de +131 a +341) se clonaron en el vector pGL3, el cual posee el gen de la luciferasa, para generar construcciones con secuencias HRE salvajes y mutadas. Se transfectaron células HeLa con estas construcciones del promotor del LRP1 (0,5μg/well) y después de 24h las células se expusieron a normoxia (barras grises) o hipoxia (barras negras) durante 24h más. Los lisados celulares se utilizaron para determinar la actividad de los promotores y se normalizaron por la actividad Renilla. Los resultados están expresados como la media±SEM de 4 experimentos realizados por triplicado (* p<0,05 vs controles). B) Las células HeLa se mantuvieron en normoxia y se transfectaron con 100ng del vector de expresión de HIF-1α (pHIF-1α) o el correspondiente vector pcADN3 vacío (como control). Después de 24h, se recogieron las células para determinar la expresión proteica y la actividad luciferasa. Los análisis de Western blot muestran la sobreexpresión de HIF-1α en las células transfectadas con el pHIF-1α vs aquellas transfectadas con el pcADN3. Los niveles de α-actina se muestran como controles de carga. C) Los lisados de las células transfectadas con pHIF-1α o pcADN3 se utilizaron para determinar la actividad luciferasa y se normalizó por Renilla. Los resultados están expresados como la media±SEM de dos experimentos realizados por triplicado (* p<0,05 vs controles en normoxia).")
Efecto de la hipoxia en la actividad del promotor LRP1. A) Diferentes regiones del promotor del LRP1 (de –1035 a +341, de –183 a +341, y de +131 a +341) se clonaron en el vector pGL3, el cual posee el gen de la luciferasa, para generar construcciones con secuencias HRE salvajes y mutadas. Se transfectaron células HeLa con estas construcciones del promotor del LRP1 (0,5μg/well) y después de 24h las células se expusieron a normoxia (barras grises) o hipoxia (barras negras) durante 24h más. Los lisados celulares se utilizaron para determinar la actividad de los promotores y se normalizaron por la actividad Renilla. Los resultados están expresados como la media±SEM de 4 experimentos realizados por triplicado (* p<0,05 vs controles). B) Las células HeLa se mantuvieron en normoxia y se transfectaron con 100ng del vector de expresión de HIF-1α (pHIF-1α) o el correspondiente vector pcADN3 vacío (como control). Después de 24h, se recogieron las células para determinar la expresión proteica y la actividad luciferasa. Los análisis de Western blot muestran la sobreexpresión de HIF-1α en las células transfectadas con el pHIF-1α vs aquellas transfectadas con el pcADN3. Los niveles de α-actina se muestran como controles de carga. C) Los lisados de las células transfectadas con pHIF-1α o pcADN3 se utilizaron para determinar la actividad luciferasa y se normalizó por Renilla. Los resultados están expresados como la media±SEM de dos experimentos realizados por triplicado (* p<0,05 vs controles en normoxia).
De acuerdo con estos resultados, la sobreexpresión de HIF-1α que da lugar a una alta acumulación de HIF-1α (fig. 4B), multiplica por 2 la actividad transcripcional del promotor salvaje del LRP1. Por el contrario, en los promotores con las cajas HRE-1 y HRE-2 mutadas, solo incrementa la actividad en 1,6 y 1,3 veces, respectivamente (fig. 4C).
Estos datos sugieren que ambas secuencias HRE juegan un papel en el efecto inductor que la hipoxia ejerce sobre el promotor del LRP1, aunque la secuencia localizada en –1072/–1069 (HRE-2) parece ser más relevante.
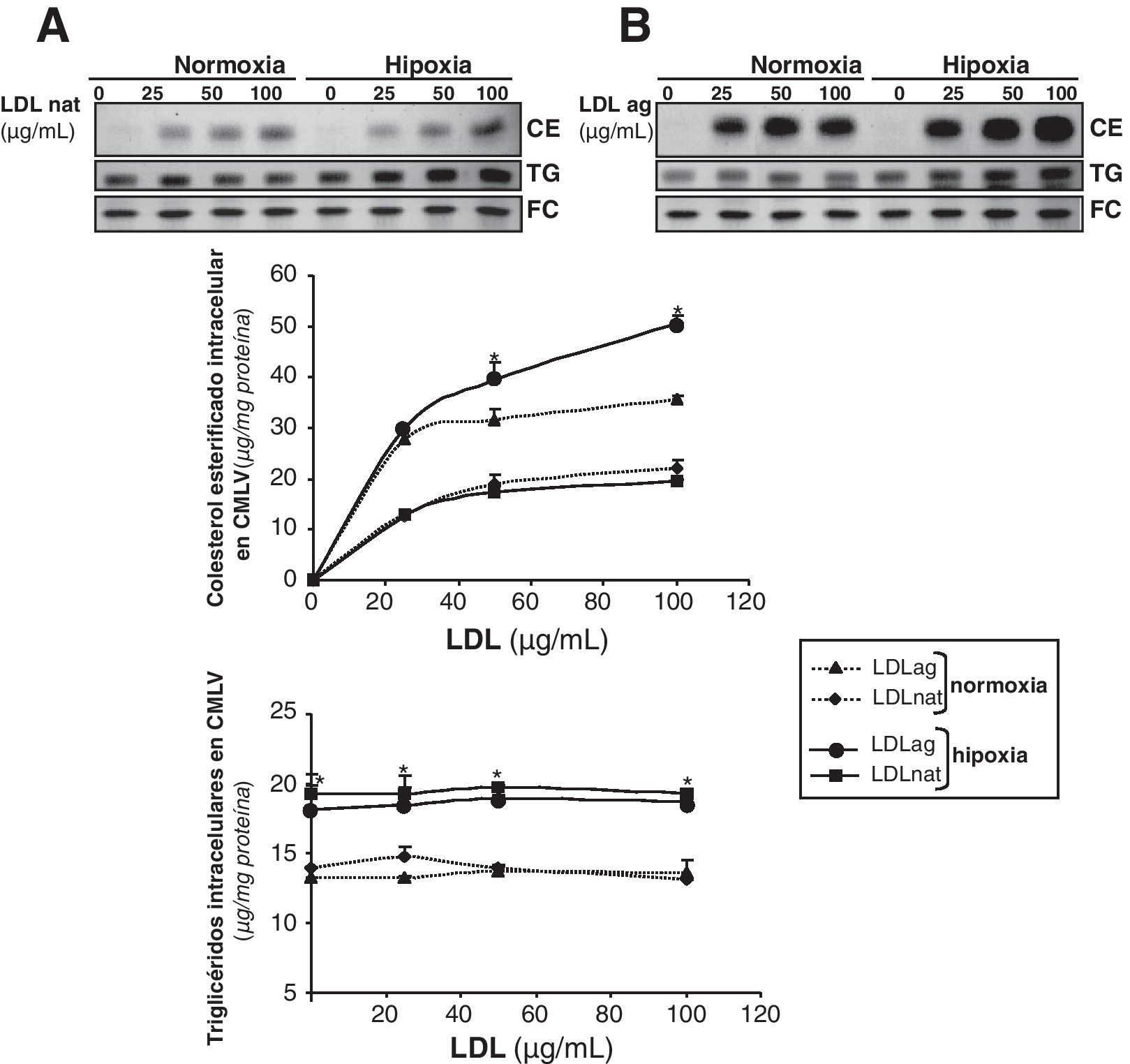
La hipoxia incrementa la captación de LDL agregadas mediada por el receptor LRP1 en CMLV humanasPara estudiar las consecuencias funcionales de la sobreexpresión del LRP1 inducida por la hipoxia, analizamos el efecto de la hipoxia en la captación de LDLag, un ligando del LRP1 en CMLV22-24. Comparamos el efecto de concentraciones crecientes de LDL nativas (LDLn) y LDLag (0, 25, 50 y 100μg/ml) en el contenido intracelular de CE de CMLV expuestas a condiciones de normoxia e hipoxia. La hipoxia incrementó la acumulación de CE derivado de LDLag (100μg/ml) desde 35,7±0,7 hasta 50,5±1,6μgCE/mg de proteína (fig. 5B). Por el contrario, las condiciones hipóxicas no alteraron significativamente la acumulación de CE derivado de LDLn a ninguna concentración (fig. 5A). El FC intracelular no varió como consecuencia del tratamiento con lipoproteínas en CMLV ni en normoxia ni en hipoxia. Sin embargo, el contenido intracelular de TG incrementó significativamente 1,5 veces en CMLV hipóxicas, independientemente de la presencia o no de LDL. Este resultado podría explicarse por el efecto, previamente descrito en macrófagos11, que la hipoxia ejerce incrementando la biosíntesis de TG.
 (A) o LDLag (B). Las células se recogieron para realizar una extracción lipídica, como se explica en la metodología. Los lípidos se separaron en una cromatografía en capa fina y se revelaron las bandas del contenido intracelular de colesterol esterificado (CE), triglicéridos (TG) y colesterol libre (FC). Los resultados están expresados como microgramos de colesterol por miligramo de proteína y se muestra la media±SEM de 3 experimentos realizados por duplicado (* p<0,05 vs controles normóxicos).")
Comparación del efecto de las LDL nativas y las LDLag en el contenido intracelular de colesterol esterificado, triglicéridos y colesterol libre en CMLV expuestas a normoxia o hipoxia. Se expusieron CMLV quiescentes a condiciones de normoxia o hipoxia durante 24h y durante las últimas 12h se incubaron con 0, 25, 50 y 100μg/ml de LDL nativas (LDLn) (A) o LDLag (B). Las células se recogieron para realizar una extracción lipídica, como se explica en la metodología. Los lípidos se separaron en una cromatografía en capa fina y se revelaron las bandas del contenido intracelular de colesterol esterificado (CE), triglicéridos (TG) y colesterol libre (FC). Los resultados están expresados como microgramos de colesterol por miligramo de proteína y se muestra la media±SEM de 3 experimentos realizados por duplicado (* p<0,05 vs controles normóxicos).
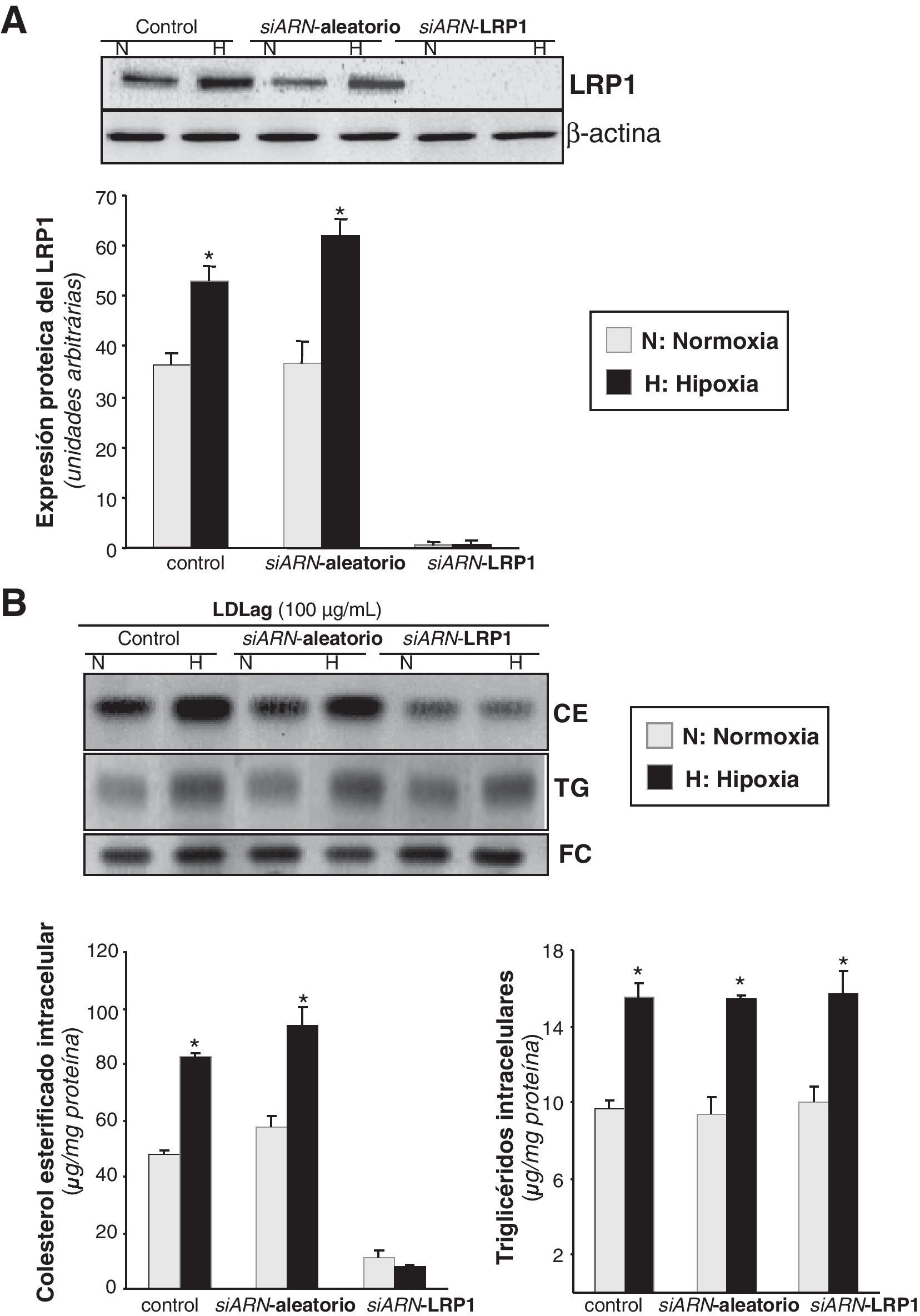
Para demostrar que el LRP1 es el responsable de la sobreacumulación de CE inducida por la hipoxia en CMLV expuestas a LDLag, comparamos el cambio en el contenido intracelular lipídico inducido por la hipoxia en células controles, nucleofectadas con un siARN aleatorio y nucleofectadas con un siARN específico para el LRP1 (siARN-LRP1). Como se muestra en la figura 6A, el LRP1 se bloqueó completamente con el tratamiento del siARN-LRP1. La hipoxia multiplicó la acumulación de CE por 1,72 en las células tratadas con el siARN aleatorio, pero no alteró la acumulación de CE en las tratadas con el siARN-LRP1 (fig. 6B). Como era de esperar, el incremento hipóxico de los TG fue similar en las células tratadas tanto con el siARN aleatorio como con el específico para el LRP1 e independientemente de la presencia o ausencia de LDLag (datos no mostrados). No se encontraron diferencias entre el contenido de colesterol intracelular de células control (nucleofectadas sin siARN) y de células tratadas con el siARN aleatorio.
 como control. También se utilizaron como control células nucleofectadas en ausencia de siARN. Después de 48h de transfección, las CMLV se expusieron a normoxia (N) o hipoxia (H) y se recogieron al cabo de 24h para evaluar la expresión del LRP1. A) El Western blot y el diagrama de barras muestran la cuantificación de la expresión proteica del LRP1. Los niveles de α-actina se muestran como controles de carga. Los resultados están expresados como la media±SEM de 2 experimentos realizados por triplicado (*p<0,05 vs controles en normoxia). B) En los experimentos de carga lipídica, CMLV quiescentes se nucleofectaron con el siARN-LRP1 o el siARN-aleatorio (0,6μmol/l) y las células control se nucleofectaron en ausencia de siARN. Después de 48h de transfección, las CMLV se sometieron a normoxia o hipoxia durante 24h, de las cuales, las últimas 12h también se incubaron con LDLag (100μg/ml). Las células se recogieron para realizar una extracción lipídica, como se explica en la metodología. Los lípidos se separaron en una cromatografía en capa fina, se revelaron las bandas del contenido intracelular de colesterol esterificado (CE), triglicéridos (TG) y colesterol libre (FC); los histogramas muestran la cuantificación de las bandas de las CMLV sometidas a normoxia o hipoxia. Los resultados están expresados como microgramos de colesterol por miligramo de proteína y se muestra la media±SEM de 3 experimentos realizados por triplicado (* p<0,05 vs controles en normoxia).")
Efecto del la inhibición del LRP1 en la sobreacumulación de colesterol esterificado derivado de LDLag inducido por la hipoxia. Las CMLV se nucleofectaron con un siARN-LRP1 o un siARN-aleatorio (0,6μmol/l) como control. También se utilizaron como control células nucleofectadas en ausencia de siARN. Después de 48h de transfección, las CMLV se expusieron a normoxia (N) o hipoxia (H) y se recogieron al cabo de 24h para evaluar la expresión del LRP1. A) El Western blot y el diagrama de barras muestran la cuantificación de la expresión proteica del LRP1. Los niveles de α-actina se muestran como controles de carga. Los resultados están expresados como la media±SEM de 2 experimentos realizados por triplicado (*p<0,05 vs controles en normoxia). B) En los experimentos de carga lipídica, CMLV quiescentes se nucleofectaron con el siARN-LRP1 o el siARN-aleatorio (0,6μmol/l) y las células control se nucleofectaron en ausencia de siARN. Después de 48h de transfección, las CMLV se sometieron a normoxia o hipoxia durante 24h, de las cuales, las últimas 12h también se incubaron con LDLag (100μg/ml). Las células se recogieron para realizar una extracción lipídica, como se explica en la metodología. Los lípidos se separaron en una cromatografía en capa fina, se revelaron las bandas del contenido intracelular de colesterol esterificado (CE), triglicéridos (TG) y colesterol libre (FC); los histogramas muestran la cuantificación de las bandas de las CMLV sometidas a normoxia o hipoxia. Los resultados están expresados como microgramos de colesterol por miligramo de proteína y se muestra la media±SEM de 3 experimentos realizados por triplicado (* p<0,05 vs controles en normoxia).
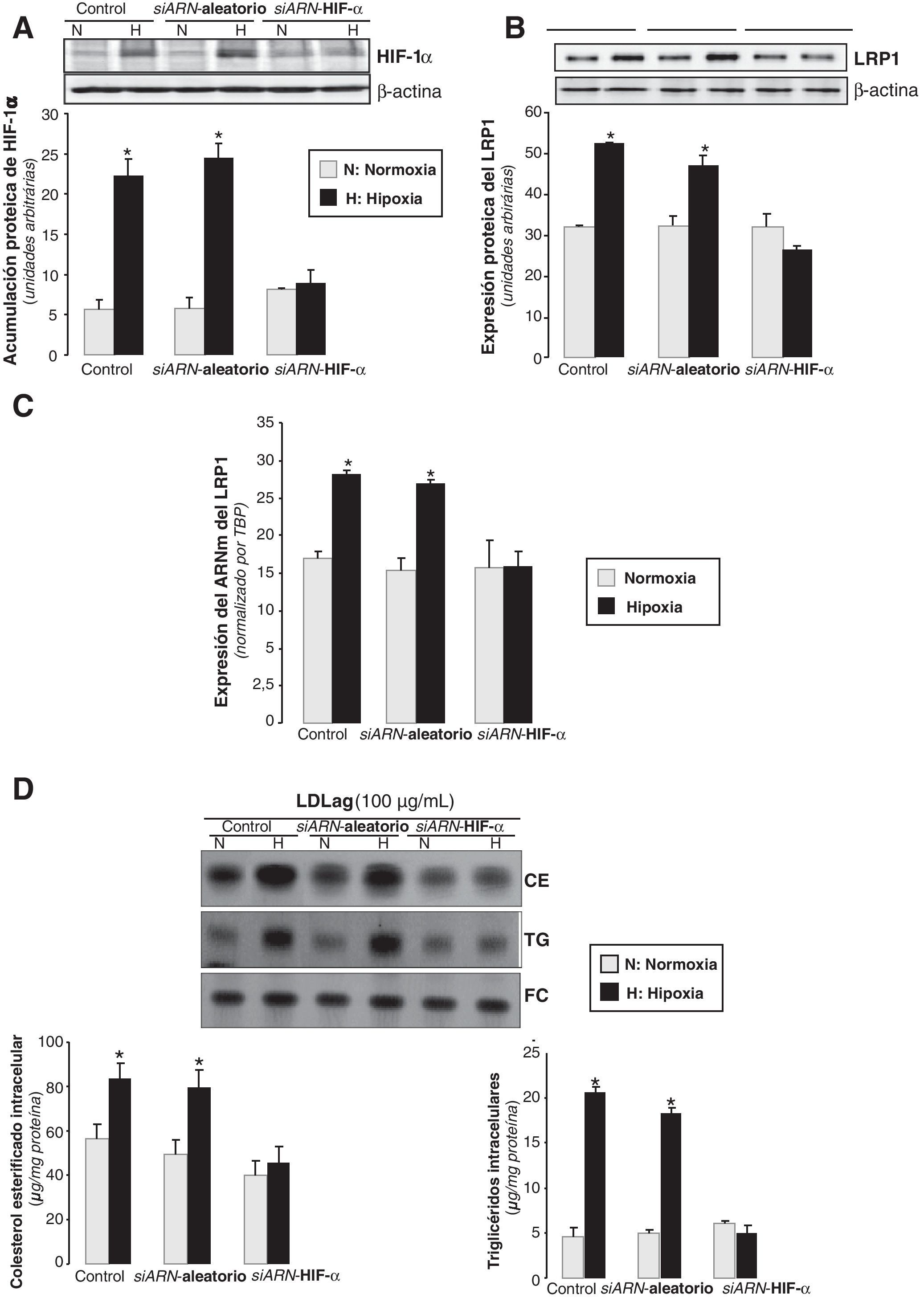
Para determinar la implicación de HIF-1α en el incremento hipóxico del LRP1, inhibimos la expresión de HIF-1α en CMLV humanas mediante la nucleofección con un siARN específico para HIF-1α (siARN-HIF-1α). El siARN-HIF-1α, pero no el siARN aleatorio, previno el incremento de los niveles proteicos de HIF-1α inducidos por la hipoxia (4,2 veces) (fig. 7A). La hipoxia no ejerció ningún efecto ni en la expresión proteica (fig. 7B) ni en la del ARNm del LRP1 (fig. 7C) en las células deficientes en HIF-1α, pero sí incrementó la expresión del LRP1 en las células controles y en las nucleofectadas con el siARN aleatorio. En concordancia, la hipoxia incrementó en 1,8 veces el contenido de CE en CMLV controles o transfectadas con el siARN aleatorio, pero no tuvo efecto en las células deficientes para HIF-1α (fig. 7D). Como era de esperar, el contenido de TG se incrementó en las células controles o transfectadas con el siARN aleatorio, pero no en las deficientes para HIF-1α.
. Las células control se nucleofectaron en ausencia de siARN. Después de 48h de transfección, las CMLV se expusieron a normoxia (N) o hipoxia (H) y se recogieron a las 2h para evaluar la acumulación de HIF-1α, o a las 18h para estudiar la expresión del LRP1 y el contenido de lípido intracelular. Está representado el Western blot y el diagrama de barras con la cuantificación proteica de HIF-1α (A) y del LRP1 (B). Los niveles de α-actina se muestran como controles de carga. Los resultados están expresados como la media±SEM de 2 experimentos realizados por triplicado (* p<0,05 vs controles normóxicos). C) Cuantificación de los niveles de expresión del ARNm del LRP1 detectados por PCR a tiempo real. Los resultados de la PCR se procesaron con un programa informático específico que utiliza los valores de los Ct normalizados por el gen endógeno (TBP). Los resultados están expresados como la media±SEM de 2 experimentos realizados por triplicado (* p<0,05 vs controles. D) En los experimentos de carga lipídica, CMLV quiescentes fueron nucleofectadas con el siARN-LRP1 o el siARN-aleatorio (0,6μmol/l). Las células control se nucleofectaron en ausencia de siARN. Después de 48h de transfección, las CMLV se sometieron a normoxia o hipoxia durante 24h, de las cuales las últimas 12h también se incubaron con LDLag (100μg/ml). Las células se recogieron para realizar una extracción lipídica, como se explica en la metodología. Los lípidos se separaron en una cromatografía en capa fina, se revelaron las bandas del contenido intracelular de colesterol esterificado (CE), triglicéridos (TG) y colesterol libre (FC); los histogramas muestran la cuantificación de las bandas. Los resultados están expresados como microgramos de colesterol por miligramo de proteína y se muestra la media±SEM de 3 experimentos realizados por triplicado (* p<0,05 vs controles en normoxia).")
Efecto de la inhibición del HIF-1α en la expresión del LRP1 inducida por la hipoxia. Las CMLV se nucleofectaron con un siARN-HIF-1α o un siARN-aleatorio (0,6μmol/l). Las células control se nucleofectaron en ausencia de siARN. Después de 48h de transfección, las CMLV se expusieron a normoxia (N) o hipoxia (H) y se recogieron a las 2h para evaluar la acumulación de HIF-1α, o a las 18h para estudiar la expresión del LRP1 y el contenido de lípido intracelular. Está representado el Western blot y el diagrama de barras con la cuantificación proteica de HIF-1α (A) y del LRP1 (B). Los niveles de α-actina se muestran como controles de carga. Los resultados están expresados como la media±SEM de 2 experimentos realizados por triplicado (* p<0,05 vs controles normóxicos). C) Cuantificación de los niveles de expresión del ARNm del LRP1 detectados por PCR a tiempo real. Los resultados de la PCR se procesaron con un programa informático específico que utiliza los valores de los Ct normalizados por el gen endógeno (TBP). Los resultados están expresados como la media±SEM de 2 experimentos realizados por triplicado (* p<0,05 vs controles. D) En los experimentos de carga lipídica, CMLV quiescentes fueron nucleofectadas con el siARN-LRP1 o el siARN-aleatorio (0,6μmol/l). Las células control se nucleofectaron en ausencia de siARN. Después de 48h de transfección, las CMLV se sometieron a normoxia o hipoxia durante 24h, de las cuales las últimas 12h también se incubaron con LDLag (100μg/ml). Las células se recogieron para realizar una extracción lipídica, como se explica en la metodología. Los lípidos se separaron en una cromatografía en capa fina, se revelaron las bandas del contenido intracelular de colesterol esterificado (CE), triglicéridos (TG) y colesterol libre (FC); los histogramas muestran la cuantificación de las bandas. Los resultados están expresados como microgramos de colesterol por miligramo de proteína y se muestra la media±SEM de 3 experimentos realizados por triplicado (* p<0,05 vs controles en normoxia).
Estos resultados demuestran que tanto el incremento del LRP1 como la sobreacumulación de CE derivado de LDLag, ambos inducidos por la hipoxia, son dependientes de HIF-1α.
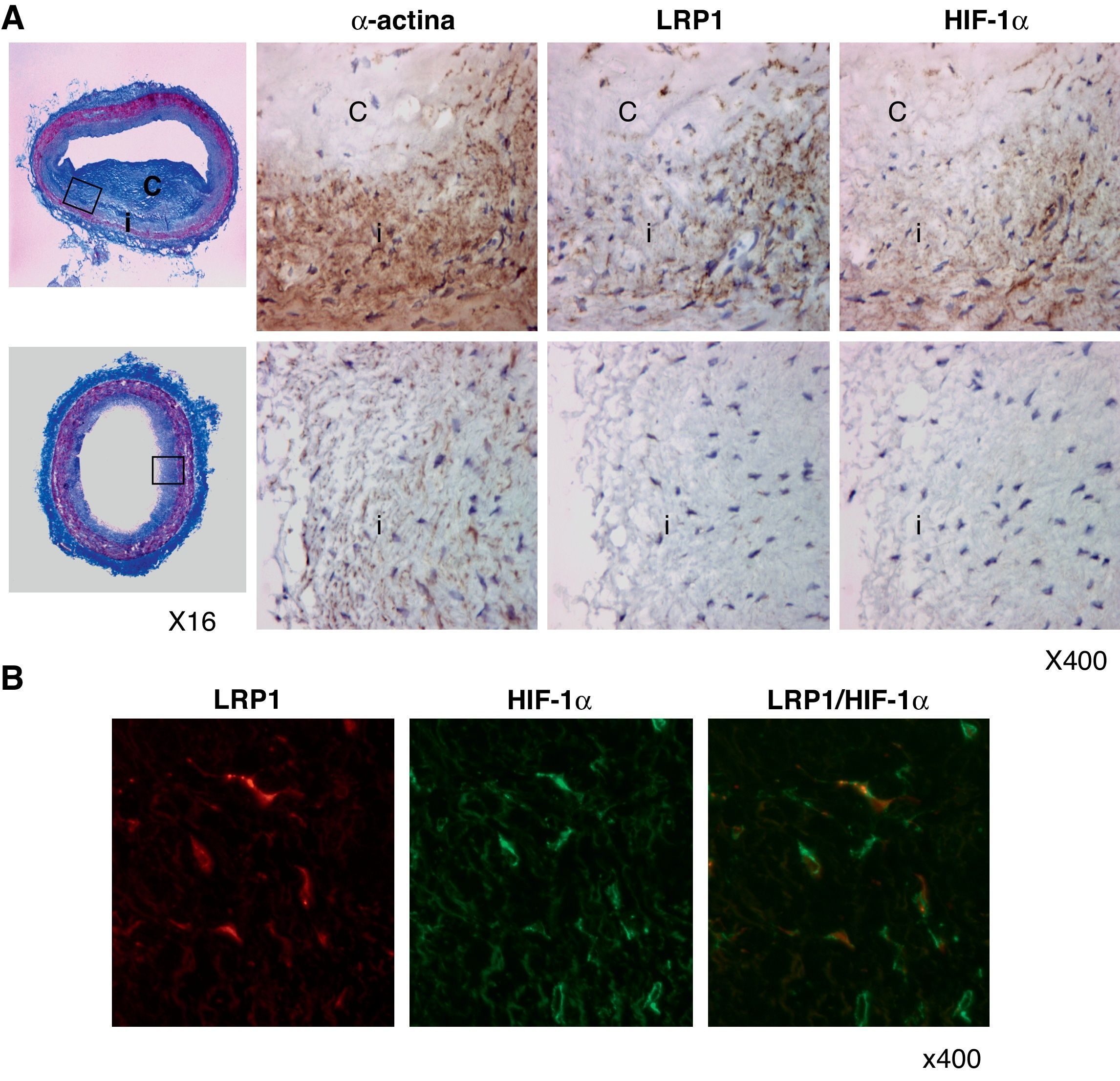
Asociación entre el LRP1 y HIF-1α en CMLV de lesiones ateroscleróticas avanzadas humanasPara saber si la regulación al alza del LRP1 por HIF-1α se da en CMLV de placas de ateroma, realizamos análisis inmunohistoquímicos. La figura 8A muestra una tinción tricrómica de Masson de una lesión aterosclerótica avanzada y de una zona normal utilizada como control. Tanto el factor de transcripción HIF-1α como el receptor LRP1 estaban altamente expresados en las zonas más profundas de la capa íntima, coincidiendo con células positivas para la α-actina (marcador específico de CMLV). Por el contrario, un leve marcaje para HIF-1α y LRP1 se observó en las zonas control. En los experimentos de doble inmunofluorescencia, el marcaje específico para el LRP1 y para el HIF-1α se solapaban en las células vasculares de las lesiones (fig. 8B).
 Tinción tricrómica de Masson de lesiones ateroscleróticas avanzadas humanas (paneles superiores) y de zonas sin lesión (paneles inferiores). Se muestra el marcaje representativo de la α-actina, el LRP1 y HIF-1α (i, íntima; c, core lipídico). Los rectángulos indican las zonas seleccionadas para el aumento. B) Las imágenes de microscopia de inmunofluorescencia muestran en amarillo la colocalización del LRP1 (en rojo) y HIF-1α (en verde) en células vasculares de lesiones ateroscleróticas avanzadas.")
HIF-1α colocaliza con el LRP1 en CMLV de lesiones ateroscleróticas humanas avanzadas. A) Tinción tricrómica de Masson de lesiones ateroscleróticas avanzadas humanas (paneles superiores) y de zonas sin lesión (paneles inferiores). Se muestra el marcaje representativo de la α-actina, el LRP1 y HIF-1α (i, íntima; c, core lipídico). Los rectángulos indican las zonas seleccionadas para el aumento. B) Las imágenes de microscopia de inmunofluorescencia muestran en amarillo la colocalización del LRP1 (en rojo) y HIF-1α (en verde) en células vasculares de lesiones ateroscleróticas avanzadas.
El LRP1 es esencial para el desarrollo embrionario40 y para el mantenimiento de la integridad vascular41. La expresión del LRP1 en la pared vascular tiene un efecto protector al limitar la señalización a través del receptor PDGF42,43. En placas ateroscleróticas ricas en lípido, el LRP1 podría contribuir a la deposición lipídica debido a su capacidad de unir e internalizar LDLag retenidas en la matriz extracelular, la principal forma de modificación de las LDL en la íntima arterial44-46. Los análisis por PCR a tiempo real y Western blot de este estudio demuestran que la hipoxia induce fuertemente la expresión del ARNm y de la proteína del LRP1 en CMLV humanas, de la misma manera que ya se había visto previamente en líneas celulares tumorales47,48. En las CMLV humanas la inducción máxima del LRP1 por la hipoxia a nivel de ARNm ocurre al cabo de 1h, y a nivel proteico a las 16h. La inducción más retardada de la proteína de este receptor por la hipoxia parece deberse a la larga vida media de la proteína del LRP1 en CMLV humanas. Los datos de nuestros experimentos con actinomicina D demuestran que la sobreexpresión del LRP1 inducida por la hipoxia depende de la transcripción, ya que la actinomicina D previene completamente este efecto de la hipoxia. La similitud entre el patrón de expresión del ARNm del LRP1 a lo largo del tiempo y el patrón de acumulación de la proteína de HIF-1α, ambos en hipoxia, apoya nuestra hipótesis de que HIF-1α modula la transcripción del promotor del LRP1. La inhibición específica de HIF-1α previene casi por completo la sobreexpresión del LRP1 inducida por la hipoxia en CMLV humanas. El análisis de la región 5’ del promotor humano del LRP1 reveló la existencia de dos posibles secuencias HRE que, de acuerdo con los ensayos de ChIP, tienen la capacidad de unir HIF-1α in vivo. La unión máxima de HIF-1α a estas secuencias coincidió en el tiempo con el máximo de acumulación de HIF-1α. Los estudios de actividad luciferasa demostraron que tanto la HRE-1 como la HRE-2 juegan un papel fundamental en la inducción del promotor LRP1 que ejerce la hipoxia y la sobreacumulación de HIF-1α, aunque la HRE-2 (–1072/–1069) parece más importante. Estos resultados concuerdan con los obtenidos en el ChIP, los cuales muestran una mayor unión in vivo de HIF-1α a la secuencia HRE-2 que a la HRE-1 en el promotor del LRP1. Estos resultados obtenidos por diferentes aproximaciones experimentales demuestran que HIF-1α media, al menos en parte, el efecto de la hipoxia sobre la transcripción del LRP1.
De forma interesante, también encontramos que la hipoxia disminuía los niveles del SREBP-1 y del SREBP-2 en las CMLV humanas. Mediante la disminución de los niveles de los SREBP, la hipoxia causó una reducción en la expresión del receptor de las LDL y probablemente esto también contribuyó a incrementar el LRP1. Anteriormente se había demostrado en nuestro grupo que de forma contraria a lo que pasaba con el receptor de las LDL, el LRP1 se regulaba negativamente por los SREBP31,32. Hay resultados controvertidos en la literatura en relación al efecto que la hipoxia ejerce sobre la expresión de los SREBP. Mientras algunos autores han publicado que se da una reducción en los niveles de los SREBP49, otros muestran resultados opuestos50,51. Estas discrepancias han sido explicadas por una modulación diferencial de los factores de transcripción por hipoxia, dependiendo del tipo celular52,53. De hecho, las diferencias en la modulación de los SREBP por la hipoxia explicaría por qué el LRP1 disminuye en CMLV cerebrales54 mientras que se incrementa en CMLV coronarias.
Nuestros resultados muestran que la inhibición de HIF-1α no solo previene la acumulación de TG inducida por la hipoxia11, sino también la sobreacumulación de CE derivado de la captación de LDLag. Por lo tanto, además de los SREBP26,31,32, HIF-1α también contribuye claramente a modular la acumulación lipídica intracelular derivada de LDLag y mediada por el LRP1 en CMLV humanas. La internalización de LDLag mediada por el LRP1 es uno de los principales mecanismos que contribuye a la formación de células espumosas derivadas de CMLV en condiciones de hiperlipidemia experimental e hipertensión22-24,26,27. De forma muy interesante, las células espumosas colocalizan con la proteína de HIF-1α en zonas con lípido de lesiones ateroscleróticas avanzadas humanas3,4. Las células espumosas no solo derivan de macrófagos, sino también de CMLV55,56. Nuestros resultados inmunohistoquímicos muestran que el LRP1 y el HIF-1α colocalizan en placas ateroscleróticas avanzadas humanas enriquecidas en lípido. El incremento en la expresión del LRP1 por un efecto combinado de la sobreacumulación de HIF-1α (inducida por la hipoxia) y la disminución de los SREBP (inducida por la presencia de lípido y por la hipoxia) podría ser relevante in vivo, ya que las zonas hipóxicas están frecuentemente asociadas con el lípido de las lesiones ateroscleróticas avanzadas3,4. El LRP1, además de ser un receptor de membrana, es un transductor de señales que se desplaza a las caveolas como respuesta a señales extracelulares57,58; de este modo, sería de interés saber si la hipoxia podría alterar la distribución subcelular del LRP1.
En resumen, los resultados de este estudio muestran que tanto HIF-1α como el LRP1 están incrementados en las CMLV hipóxicas, y que el marcaje proteico de HIF-1α y del LRP1 coinciden en CMLV de placas de ateroma avanzadas. Todos estos resultados de forma conjunta demuestran que las CMLV hipóxicas podrían contribuir a los elevados niveles de HIF-1α asociados a las células espumosas en lesiones ateroscleróticas avanzadas humanas3,4.
FinanciaciónEste trabajo se hizo posible gracias a la financiación del CIBEROBN03/2006, REDINSCOR RD06/0003/0015 y FIS PI080351 del Instituto Salud Carlos III, cofinanciado por el Fondo Europeo de Desarrollo Regional (F.E.D.E.R.), MARATÓ TV3-Malalties Cardiovasculars, SAF 2006/10009, Fundación Lilly y FIC-F.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
AgradecimientosLos autores quieren dar las gracias al equipo de trasplantes de corazón del Servicio de Cardiología y Cirugía Cardiaca del Hospital de la Santa Creu i Sant Pau en Barcelona. También agradecemos el apoyo técnico de Laura Nasarre y Marta Sánchez. L.B., V.LL.-C. y J.C. son miembros del programa de graduación internacional PROMISE (IRTG-1566), apoyado por el Deutsche Forschungsgemeinschaft (Bonn, Alemania).