




El receptor activado por proliferadores peroxisómicos tipo alfa (PPARα) une ácidos grasos, eicosanoides o fibratos, y regula la transcripción de genes relacionados tanto con el metabolismo lipídico y glucídico como con la inflamación. Los fibratos son ligandos de PPARα que se utilizan para normalizar diversos parámetros lipídicos y glucídicos y que ejercen efectos antiinflamatorios. De hecho, se ha demostrado que los fibratos producen mejoras en los pacientes con síndrome metabólico, diabetes mellitus de tipo 2 y enfermedades cardiovasculares. Este artículo revisa el mecanismo de acción y los papeles funcionales de los fibratos, enfatizando los factores que modulan su capacidad para activar PPARα y que por tanto pueden afectar su efectividad. Estos factores podrían explicar los resultados obtenidos en experimentos con animales y en estudios clínicos con fibratos en los que se encontraron efectos adversos y/o una ineficiente acción hipolipemiante tras la activación del PPARα. Por lo tanto, todos esos resultados han subrayado la necesidad de buscar otros fármacos o agentes nutricionales que modulen el PPARα. Esta estrategia podría ser complementaria o bien reemplazar al tratamiento clásico. Así, revisamos brevemente los nuevos ligandos naturales y sintéticos del PPARα que se están desarrollando actualmente y que son más específicos, selectivos, con más potencia y que, en principio, presentan mayor efectividad y menos efectos adversos que los fibratos.
Peroxisome proliferator-activated receptor alpha (PPARα) binds fatty acids, eicosanoids, or fibrates, and regulates transcription of genes involved both in lipid and glucose metabolism as well as in inflammation. Fibrates are PPARα ligands used to normalize lipid and glucose parameters and exert anti-inflammatory effects. In fact, fibrates have already been demonstrated to benefit metabolic syndrome, type 2 diabetes and cardiovascular diseases. This article reviews the mechanism of action and the functional roles of fibrates, emphasising the factors modulating their capacity to activate PPARα and affecting their effectiveness. These factors would possibly explain the findings obtained in animal studies and clinical trials with fibrates which showed either untoward effects and/or inefficient hypolipidemic action of PPARα activation. Therefore, all those findings have underlined the need to search for pharmacological or nutritional solutions based on modulation of PPARα. This strategy could either be complementary to or replace the classical therapy. Thus, a brief survey of the natural and synthetic agonists of PPARα, more specific, selective, with higher potency and, supposedly, displaying greater effectiveness and fewer adverse effects than fibrates which are currently being developed, is also mentioned.
Los receptores activados por proliferadores peroxisómicos (PPAR) pertenecen a la superfamilia de receptores nucleares de hormonas, junto con los receptores de hormona tiroidea, retinoides, hormonas esteroideas y de vitamina D1. Los PPAR son factores de transcripción dependientes de ligando que regulan diversos aspectos de la homeostasia energética, metabolismo lipídico y lipoproteico, homeostasia glucídica, metabolismo de aminoácidos y de la síntesis de urea. Este hecho indica que los PPAR pueden funcionar como una molécula clave implicada en diversas alteraciones metabólicas. Es por ello que hay un interés cada vez mayor en la búsqueda de soluciones terapéuticas que utilicen como diana los PPAR, tanto en forma de agentes nutricionales como farmacológicos. En los últimos años, los PPAR han cobrado también importancia como reguladores de la inflamación y la respuesta immunitaria, y han abierto una nueva área de desarrollo de agentes terapéuticos que se emplean en el tratamiento de enfermedades que cursan con inflamación crónica, como aterosclerosis, resistencia insulínica inducida por la obesidad y enfermedades neurodegenerativas.
En este trabajo, se revisa el mecanismo de activación del PPARα, y se analizan algunos casos en los cuales los agonistas del PPARα no han obtenido la efectividad deseada o bien han provocado efectos adversos. También se discute el empleo de nuevos fármacos o nutrientes que pudieran prevenir o aliviar los efectos inesperados producidos por agonistas del PPARα, que hemos revisado recientemente2.
Características estruturalesEn 1990, los PPAR se identificaron como receptores para los fibratos, un tipo de fármacos hipolipemiantes empleados en humanos desde finales de los años sesenta3. Debido a su capacidad de inducir la proliferación de peroxisomas en roedores recibieron el nombre de PPAR4.
Los PPAR son proteínas de 49–56kDa, con una estructura modular que consta de 6 dominios funcionales, definidos como A/B, C, D y E/F. El dominio aminoterminal A/B contiene una función de transactivación independiente de ligando (AF-1)5. El dominio C o dominio de unión al ácido desoxirribonucleico (ADN) (DBD) se encuentra estabilizado por 2 átomos de cinc unidos cada uno a 4 residuos de cisteína. Este dominio DBD está involucrado en la heterodimerización con el receptor X del ácido 9-cis retinoico (RXR) (NR2B), y forma un complejo que es capaz de unirse al elemento respuesta a proliferadores peroxisómicos (PPRE), que se localiza en el promotor de los genes diana del PPAR6. El dominio D o dominio bisagra une el dominio DBD y el dominio de unión al ligando (LBD), lo que permite doblar o alterar conformacionalmente la proteína. El dominio D permite el acoplamiento de cofactores, está involucrado en la localización nuclear del receptor y contiene un sitio de fosforilación para quinasas7. Finalmente, el dominio E/F o LBD, localizado en el carboxilo terminal, contiene un bolsillo hidrofóbico que presenta 2 diferencias respecto a otros receptores nucleares: una enorme cavidad de unión al ligando y una hélice α adicional en la parte inferior del bolsillo que participa en la entrada de los ligandos8. Estas características pueden contribuir a la capacidad de los PPAR de unirse a un amplio rango de compuestos lipofílicos, tanto sintéticos como naturales. El dominio E/F también contiene una función de transactivación dependiente de ligando (AF-2) y presenta una serie de regiones que permiten la interacción con RXR, cofactores y proteínas de choque térmico9.
Isotipos, distribución tisular y ligandosSe han identificado 3 isoformas: PPARα (NR1C1), PPARβ (llamado también −δ, NR1C2), y PPARγ (NR1C3)10, que presentan distinta distribución tisular, lo que refleja sus diferentes funciones biológicas. Los patrones de expresión no difieren significativamente entre machos y hembras11. La expresión del PPARα es más elevada en tejidos con un activo catabolismo de los ácidos grasos, como el hígado, el riñón, el corazón, el músculo esquelético y el duodeno11. Además, los PPAR se expresan en diferentes tipos de células vasculares e inmunitarias, como monocitos/macrófagos, células endoteliales (EC), células de la musculatura lisa vascular (VSMC), linfocitos y células dendríticas.
A pesar de una estructura general común del LBD, la unión de algunos ligandos a PPARα, β y γ muestra especificidad tanto de especie, como de isotipo. Los PPAR se activan mediante compuestos naturales y sintéticos.
Ligandos naturalesLa identificación de los ácidos grasos insaturados como ligandos del PPAR prueba, al menos en parte, la firme evidencia de que la actividad transcripcional dependiente de PPAR de los ácidos grasos es el resultado de una interacción directa con este receptor nuclear. En general, todas las isoformas de PPAR presentan una selectividad mayor por los ácidos grasos poliinsaturados de cadena larga (PUFA) omega 3 (ω-3) y ω-6 que por los ácidos grasos saturados o monoinsaturados12. Estos ácidos grasos se unen a las 3 isoformas, aunque presentan más afinidad por el PPARα. Los ácidos grasos saturados son ligandos débiles del PPAR, mientras que los ácidos grasos ramificados y los derivados de isoprenoides se unen de forma eficaz al PPARα12. De acuerdo con esto, compuestos relacionados con procesos inflamatorios, como eicosanoides derivados del ácido araquidónico vía lipooxigenasa12, leucotrienos (LT) y ácidos grasos oxidados, son también ligandos naturales del PPARα13.
Ligandos sintéticosLos PPAR son también activados por proliferadores peroxisómicos, una clase amplia de compuestos estructuralmente diversos que incluyen fármacos hipolipemiantes, agentes plastificantes, herbicidas y solventes. Estos agenes hipolipemiantes son del tipo fibratos, como clofibrato, fenofibrato, bezafibrato, gemfibrocilo y un compuesto experimental, Wy-14,643. Los fibratos se unen preferencialmente al PPARα4, mientras que las tiazolidinedionas (TZD), una clase de fármacos antidiabéticos, se unen selectivamente al PPARγ14. Entre los compuestos sintéticos que se unen al PPARα se incluyen al 5,8,11,14 ácido eicosatetrainoico (ETYA), un análogo del ácido araquidónico15, y agentes antiinflamatorios no esteroideos (NSAID), que también parecen activar al PPARγ16. También parece ser que agonistas del RXR, como el ligando natural ácido 9-cis retinoico, puede inducir la formación del heterodímero PPAR:RXR y activar genes diana del PPAR17.
Mecanismo de acciónLas actividades biológicas y terapéuticas del PPAR son el resultado de la combinación de 2 mecanismos de actuación diferentes (Fig. 1): a) una transactivación dependiente de ligando encargado de controlar los efectos metabólicos, y b) una transrepresión dependiente de ligando, que controla los efectos vasculares. Otros mecanismos importantes en la activación del PPAR son: su contenido celular, la naturaleza de los ligandos, la señal cruzada con otros receptores nucleares y la estructura del PPRE. Además, otros mecanismos de control a tener en cuenta son las modificaciones postraduccionales que puede presentar el PPAR, como fosforilación, sumoilación y ubiquitinación, entre otrosbb0090, bb0095.
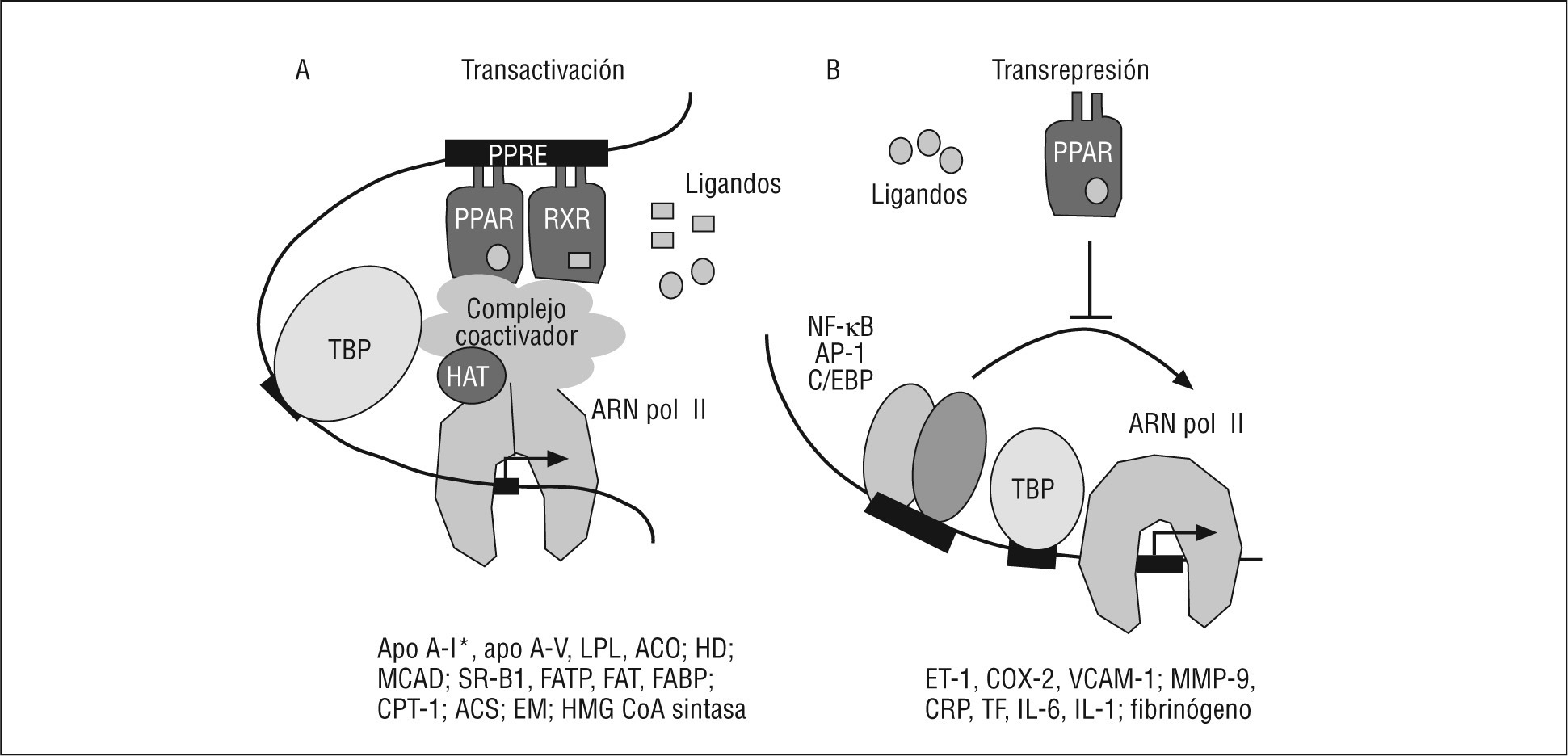
Fig. 1. Mecanismos de señalización del receptor activado por proliferadores peroxisómicos tipo alfa (PPARα). A. Transactivación: el PPARα regula la transcripción de sus genes diana por heterodimerización con el receptor X del ácido 9-cis retinoico (RXR). El hetodímero PPARα/RXR se une al elemento respuesta a proliferadores peroxisómicos (PPRE) localizado en el promotor de sus genes diana. El heterodímero PPARα/RXR, una vez activado por el ligando, se asocia con cofactores que presentan actividad acetiltransferasa de histonas (HAT), lo que modifica la estructura del nucleosoma, posibilita el contacto con los factores de transcripción generales y permite la transcripción del gen. Se citan algunos genes diana del PPARα, implicados en el metabolismo lipídico. B. Transrepresión: el PPARα puede reprimir la transcripción de genes a través de mecanismos diversos (ver el texto) que son independientes de su unión al ácido desoxirribonucleico. Esta represión puede deberse a la interferencia del PPARα en la actividad de otros factores de transcripción, implicados en la modulación de la expresión de genes generalmente relacionados con inflamación.ACO: acil-CoA oxidasa; AP-1: activador de la proteína 1; Apo: apolipoproteína; ARN pol II: ácido ribonucleico polimerasa tipo II; C/EBP: proteína de unión al intensificador/CCAAT; COX-2: ciclooxigenasa 2; CPT-1: carnitina palmitoil transferasa 1; EM: enzima málica; ET-1: endotelina 1; FABP: proteína de unión a los ácidos grasos; FAT: transportador de ácidos grasos; FATP: proteína transportadora de ácidos grasos; HD: hidratasa deshidrogenasa; IL: interleucina; LPL: lipoproteína lipasa; MCAD: acil-CoA deshidrogenasa de cadena media; MMP-9: metaloproteinasa de la matriz 9; NF-κB: factor nuclear-kappa B; PCR: proteína C reactiva; SR-B1: receptor scavenger de tipo B1; TBP: proteína de unión a la caja TATA (TATA binding protein); TF: factor tisular; VCAM-1: molécula de adhesión celular vascular 1.*Presentan un efecto diferente en humanos y roedores.
Activación transcripcionalLa funcion típica del PPAR es la activación dependiente de ligando de la transcripción, después de haberse unido directamente al ADN a través del elemento respuesta del promotor o intensificador (enhancer) de los genes diana. La regulación transcripcional de los PPAR requiere la heterodimerización con el RXR, el cual pertenece a la misma superfamilia de receptores. El complejo PPAR: RXR, activado por ligando, modula la transcripción mediante la unión a una secuencia específica del ADN, denominada PPRE (Fig. 1). Este elemento respuesta está compuesto generalmente por una secuencia hexanucleotídica (AGGTCA) que se repite de manera directa con una separación entre las 2 copias de tan sólo 1 nucleótido. De ahí que se denomine DR-1. No obstante, la capacidad de los receptores nucleares de iniciar o suprimir el proceso de transcripción depende de su interacción con cofactores negativos o positivos. Estos cofactores sirven como puente entre los factores de transcripción y la maquinaria transcripcional básica (Fig. 1) y, lo más importante, estos cofactores contienen ciertas actividades enzimáticas que controlan la expresión de los genes mediante la modificación específica de la estructura del ADN y de la cromatina. La acetilación de histonas es uno de estos mecanismos críticos que regulan la expresión de genes. Por regla general, un aumento en los valores de acetilación de histonas relaja el empaquetamiento de la cromatina, lo que se relaciona con una activación transcripcional mayor, mientras que una disminución en la actividad de histona acetilasa se asocia con una represión transcripcional20. De acuerdo con esto, la proteína de unión al CREB (proteína de unión al elemento respuesta de AMP cíclico) o CBP y la p300 son coactivadores21 que presentan actividad histona acetilasa22, mientras que los 2 correpresores, mediador de silenciamiento para receptores de retinoides y hormona tiroidea (SMRT, del inglés silencing mediator for retinoid and thyroid hormone receptor), y el correpresor del receptor nuclear (NCoR), contienen actividad desacetiladora de histonas23. Así, en ausencia de ligando, los heterodímeros PPAR:RXR se asocian con correpresores, como NCoR y SMRT24. El estado desacetilado de histonas inhibe la transcripción25. Por el contrario, la unión al ligando induce cambios conformacionales en el dominio de transactivación (AF-2) del LBD del PPAR, lo que provoca la liberación de los correpresores y la selección de coactivadores26. Numerosos coactivadores, como el PBP (proteína de unión al PPAR), PRIP/RAP250 (proteína de interacción con PPAR), PGC-1 (coactivador 1 del PPAR gamma) o el coactivador del receptor esteroideo 1 (SRC-1), son capaces de interaccionar con PPAR in vitro. La interacción de receptores nucleares con estos coactivadores genera la unión del heterodímero al PPRE en el promotor, la modificación de la estructura de la cromatina y la activación o supresión de la transcripción del gen diana. Este modelo implica que la cromatina tiene que ser lo suficientemente flexible para permitir la formación de un bucle (Fig. 1).
Represión génicaEl PPAR también puede regular negativamente, de manera dependiente del ligando, la expresión de genes, mediante la inhibición de la actividad de otros factores de transcripción, como los miembros de las familias del factor nuclear-kappa B (NF-κB) y del activador de la proteína 1 (AP-1)bb0135, bb0140, el transductor de señal y activador de la transcripción (STAT), la proteína de unión al intensificador/CCAAT (C/EBP)18 y el factor nuclear hepático 4 alfa (HNF4α), entre otros29.
En este mecanismo no es necesaria la unión del receptor al elemen to respuesta específico en la molécula de ADN (Fig. 1). Sin embargo, a pesar de la gran cantidad de estudios realizados, los mecanismos por los cuales los PPAR inhiben estas rutas de transducción de señales no están completamente entendidos. Recientemente, Ricote et al30 han revisado los mecanismos moleculares que pueden explicar la transrepresión específica de genes mediada por ligandos del PPAR. Entre ellos se encuentran las interacciones directas proteína:proteína entre el PPAR y otros factores de transcripción, la modulación de la actividad cinasa, la competición por los coactivadores y el modelo dependiente del correpresor. Más aún, uno de estos modelos de transrepresión establece que el PPARα regula positivamente la expresión del represor del NF-κB llamado inhibidor de kappa B (IκB)31, que actúa como secuestrador de subunidades del NF-κB, impidiendo tanto su translocación al núcleo como, consecuentemente, su capacidad de unión al ADN32. Por lo tanto, el PPARα puede inhibir genes inducidos por NF-κB. Por otro lado, la interferencia del PPARα con la señal del C/EBP y del HNF4α podría explicarse porque el PPARα es capaz de disminuir la expresión del HNF4α33, así como la del propio C/EBP34 (Fig. 1).
Mecanismos que controlan la actividad del PPARαDado que el presente artículo está enfocado principalmente al PPARα, vamos a analizar el mecanismo de acción exclusivamente de este subtipo.
Contenido celular relativo del PPARαEn cuanto a la expresión del PPARα, algunos estudios muestran una regulación de esta expresión por glucocorticoides35, hormona de crecimiento36, insulina o, incluso, por ácidos grasos y fibratosbb0185, bb0190. Así pues, se ha descrito que los valores de ácido ribonucleico (ARN) mensajero (ARNm) y proteína de PPARα discurren en paralelo con el ritmo circadiano de los valores circulantes de glucocorticoides. A su vez, situaciones de estrés o ayuno que producen un aumento en los valores plasmáticos de glucocorticoides también provocan un incremento en la síntesis de PPARα35. Por el contrario, la hormona de crecimiento parece que disminuye los valores de ARNm del PPARα36. Por otro lado, hay resultados contradictorios sobre la regulación de la expresión del PPARα por la insulinabb0195, bb0200, aunque parece cierto que el efecto inhibidor de la insulina sólo se observa cuando la expresión del PPARα está aumentada. Este efecto contrarregulador de la insulina se ha observado tanto in vitro por Steineger et al40, como in vivo por nosotros37. Nuestros estudios in vivo con ratas han mostrado que durante el desarrollo perinatal, los valores de ARNm y proteína PPARα varían marcadamente, y presentan un máximo durante el período de lactancia, seguido de un pronunciado descenso cuando las ratas se hacen adultas41. Esta diferencia está claramente relacionada con la ingestión de leche, rica en lípidos, y con la insulinemia baja que concurren durante el período de lactancia37. De hecho, se pudo comprobar que una sobrecarga oral de grasa era capaz de incrementar de forma acusada la expresión hepática de PPARα, a menos que los valores plasmáticos de insulina fueran elevados37. Por último, un aumento en la expresión del gen del PPARα mediada por sus propios ligandos, fibratos y ácidos grasos libres (FFA), sugeriría una autorregulación de este receptor nuclear. Esta situación se ha observado tanto en células de ratabb0195, bb0210, como en estudios in vivo en hígado de rata38, y se ha confirmado en ratones carentes de PPAR33.
Es interesante señalar que los valores de expresión del PPARα determinan su respuesta a los agonistas. Duez et al43, utilizando ratones transgénicos PPARα+/− con apolipoproteína (apo) A-I humana, demostraron que los fibratos son menos efectivos en la reducción de los lípidos plasmáticos y en la modulación de los valores de ARNm de los genes diana si se comparaban con ratones PPARα+/+ con apo A-I humana. Estos hallazgos muestran que el PPARα influye en la respuesta a los fibratos de una manera dependiente de la dosis de gen del propio receptor que esté presente43. Ello concuerda con la hipótesis de Holden y Tugwood44, los cuales propusieron que los valores de expresión del PPARα pueden mediar diferentes respuestas a los agonistas del PPARα en la expresión de los genes. De este modo, mientras que valores altos de PPARα podrían modular tanto la expresión de genes involucrados en la homeostasia lipídica como la de genes relacionados con la proliferación peroxisómica, cantidades bajas del PPARα podrían actuar sólo en la regulación de los primeros44. Una explicación así podría justificar la sensibilidad diferente encontrada por nosotros en la expresión de genes de la proliferación peroxisómica (el propio PPARα y acil-CoA oxidasa [ACO]) y de genes de la homeostasia lipídica y glucídica (fosfoenolpiruvato carboxiquinasa [PEPCK] y lipoproteína lipasa [LPL]) tras cambios nutricionales, en el hígado de ratas lactantes (que presentan valores altos de PPARα) y en ratas adultas (que presentan valores bajos de PPARα)45. De forma similar, permitiría explicar por qué, en humanos, sólo se inducen tras la exposición a ligandos del PPARα los genes relacionados con la hipolipidemia y, por el contrario, en roedores, se activan tanto los genes asociados con el metabolismo lipídico como los genes relacionados con la proliferación peroxisomal46. De hecho, se ha indicado que los menores valores de expresión del PPARα detectados en el hígado humano, en comparación con los del hígado de ratón, podrían ser la causa de la falta de respuesta de los humanos a la proliferación peroxisomal47. Así pues, en roedores, los agonistas del PPARα inducen mucho más la β-oxidación peroxisómica que la ruta mitocondrial, y por el contrario en humanos, la β-oxidación mitocondrial es fuertemente inducida, mientras que la ruta peroxisómica no es afectada. Relacionado con todo esto, se ha observado que el tratamiento crónico con proliferadores peroxisómicos provoca hepatocarcinoma en roedores, mientras que los estudios epidemiológicos no han mostrado este efecto en pacientes tratados con fibratos46. Aún más, los activadores de PPARα pueden inducir proliferación peroxisómica también en el corazón del ratón, lo que podría incrementar el estrés oxidativo48, resultando ser cardiotóxico49, y provocando hipertrofia y fallo cardíacos50. No obstante, hasta ahora no se ha encontrado que en humanos el tratamiento con fibratos predisponga a una insuficiencia cardíaca crónica.
Otro mecanismo potencial que podría producir cambios a largo plazo en la expresión de genes es una metilación alterada del ADN. La metilación en la posición 5’ de residuos de citosina es una modificación común en el genoma de los mamíferos y se asocia con la represión transcripcional, mientras que la hipometilación se encuentra asociada con una actividad transcripcional elevada51. Es interesante resaltar que estas alteraciones en el fenotipo provocan mecanismos moleculares denominados epigenéticos, que implican cambios estables en la expresión génica, lo que ha provocado un interés creciente en su estudio. Es más, se ha visto que diversas alteraciones que ocurren durante la vida materna pueden provocar una programación fetal que refleje una susceptibilidad aumentada a presentar enfermedades posnatales y que afectan a la impronta genómica52. Por ejemplo, se ha visto que la restricción proteica en la dieta de la madre durante la gestación en ratas altera la expresión del PPAR y de sus genes diana, y se ha relacionado con una homeostasia lipídica peor en la descendencia. Una menor metilación del promotor del PPARα se ha asociado con una expresión mayor del propio ARNm51. Además, recientemente se ha observado que la exposición a agonistas del PPARα también produce efectos epigenéticos en el hígado de ratón y que son dependientes de la presencia del PPARα53.
El contenido nuclear del PPARα podría ser también determinante en su capacidad de regulación de la expresión de genes. La forma libre (no unida a ligando) del PPARα se encuentra generalmente presente en el citosol y forma un complejo con chaperonas citoplasmáticas. De hecho, se ha observado que los PPAR son capaces de unirse a proteínas de choque térmico (hsp, del inglés heat-shock proteins) tanto a la de 72-kDa54 como a la hsp909. Tras su unión a un agonista, el complejo se disocia y el PPARα migra al núcleo55.
Finalmente, los valores de receptor nuclear pueden controlarse también por la degradación y la estabilidad de la proteína. Recientemente, se ha demostrado que numerosos receptores nucleares se degradan por la vía de la ubiquitina-proteosoma. Las proteínas degradadas por esta vía se modifican covalentemente por fijación de un polipéptido, llamado ubiquitina, en residuos de lisina. La proteína del PPARα está generalmente ubiquitinada y sus ligandos son capaces de disminuir la ubiquitinación del receptor nuclear, o, lo que es lo mismo, un mecanismo de estabilización de la proteína dependiente de ligando56. Además, la interacción del PPARα con RXRα o CBP conduce a una reducción en la vida media de la proteína. Por el contrario, la interacción con NCoR, el cual inhibe su actividad transcripcional, conlleva una estabilización de la proteína57.
Naturaleza del ligandoEl dominio LBD del PPARα presenta una gran cavidad que permite acomodar muchos tipos diferentes de ligandos naturales y sintéticos48.
Una amplia variedad de ácidos grasos saturados e insaturados puede unirse al PPARα, aunque con una baja afinidad. No obstante, los cambios inducidos por la dieta en los valores plasmáticos de ácidos grasos insaturados pueden llegar a ser suficientes para modificar ligeramente la actividad del PPAR18. Debido a la cantidad relativamente alta de posibles ligandos endógenos que están presen-tes en la célula, es difícil establecer qué molécula es el verdadero ligando endógeno fisiológico del PPARα48. Por tanto, una estrategia alternativa ha sido evaluar rutas enzimáticas que pudieran generar ligandos de manera local. De este modo, se ha demostrado que la LPL libera ácidos grasos desde las lipoproteínas ricas en triglicéridos, que son capaces de activar al PPARαbb0290, bb0295. Sin embargo, al utilizar otras lipasas que eran igual de eficaces en generar FFA, no se conseguía activar al PPARα, lo que indica diferencias en la localización intracelular de esos ácidos grasos o bien una regulación selectiva del receptorbb0290, bb0300. En este mismo sentido, parece ser que los ácidos grasos sintetizados de novo son capaces de regular la actividad del PPARα, mientras que los ácidos grasos liberados desde el adipocito resultan inactivos. Una posibilidad es que las proteínas de unión a los lípidos seleccionen los ácidos grasos que van a ser transportados hasta el PPARα61. Se ha indicado un posible papel de la proteína de unión a los ácidos grasos (FABP) y de otras proteínas transportadoras citoplasmáticas, para guiar a los ligandos hasta los receptores nucleares y, así, mediar en la interacción del PPAR con sus agonistasbb0310, bb0315, bb0320. De hecho, recientemente se ha confirmado un papel así para el ácido retinoico, sus proteínas de unión y sus receptores nucleares65.
De manera similar a lo observado para los ligandos naturales, los fibratos que son ligandos de afinidad baja para el PPARα deben usarse generalmente a dosis altas para alcanzar una actividad hipolipemiante eficaz66. Además, los fibratos parecen actuar preferencialmente en el hígado, mientras que otros ligandos de más afinidad, como Wy-14,643 y GW7647, actúan de manera más eficaz en tejidos extrahepáticosbb0240, bb0300, indicando que hay una especificidad tisular en la activación del PPARα67. Para hacerlo más complejo, no se pueden descartar diferencias interespecie en la activación del PPARα por sus ligandos. Así, por ejemplo, los fibratos son más efectivos que el ácido linoleico en activar al PPARα en células en cultivo de hepatoma de rata, mientras que el linoleico es más efectivo que los fibratos en células de hepatoma humano68.
Es interesante señalar que se ha comprobado que los ácidos grasos de cadena larga pueden competir o desplazar a los proliferadores peroxisómicos en su unión a FABP62, lo cual dependerá de la concentración intracelular relativa de los 2 tipos de agonistas de PPARα. Así pues, en experimentos libres de células, se ha observado que los ácidos grasos liberados en la hidrólisis catalizada por la LPL de las lipoproteínas de muy baja densidad (VLDL) son capaces de desplazar a potentes ligandos sintéticos de unirse a la proteína PPARα58. De acuerdo con todo esto, en nuestros estudios realizados con ratas tratadas con fenofibrato al final de la gestación, podría estar ocurriendo una posible competición entre el fibrato y los FFA, cuyos valores en plasma se sabe que son elevados en esta etapa de la gestación, de manera que ambos agonistas estarían regulando la expresión de los mismos genes, pero también de genes diferentes, lo que provocaría respuestas biológicas tanto específicas como comunesbb0190, bb0345. Esto también ocurriría en el caso de animales que han recibido una dieta grasa junto con fibratos67. Se ha indicado que cada uno de estos ligandos del PPARα podría tener una afinidad de unión distinta, con un tiempo de residencia en el receptor diferente, y con una forma de interaccionar con la cavidad de unión desigual. Por ello, los FFA podrían disminuir la accesibilidad de los fibratos a su correspondiente sitio de unión al PPAR, reducir sustancialmente la capacidad de los fibratos de activar al PPARα y, consecuentemente, sus efectos metabólicos. Es más, se ha indicado que la modulación de la expresión de diversos genes diana por fibratos y por ácidos grasos a menudo está desconectada. De hecho, estas y otras respuestas diferentes indicarían que no todos los efectos provocados por los ácidos grasos se deben achacar a la activación del PPAR70.
Reacción hormonal cruzada con otros receptores nuclearesLos elementos DR-1 presentes en el PPRE son también reconocidos por otros receptores nucleares, como RAR:RXR, homodímeros de RXR, HNF-4 y factor de transcripción de la ovoalbúmina de pollo (COUP-TF). De hecho, los homodímeros, tanto de HNF-4 como de COUP-TF, pueden desplazar al PPAR:RXR de su sitio de unión y competir con la señal del PPARbb0355, bb0360. La eficacia de estas interacciones depende de la secuencia del elemento regulador y del contexto del promotor, lo que indica una reacción cruzada con otros receptores nucleares que podría generar acciones de tipo agonista o antagonista e influir en el control metabólicobb0240, bb0365. Por otra parte, el RXR es la pareja indispensable en la dimerización del PPAR y de otros receptores nucleares, como RAR, receptor de la hormona tiroidea (TR), receptor de la vitamina D (VDR), entre otros. Además, como se mencionó anteriormente, hay múltiples tipos de cofactores (coactivadores y/o correpresores) que los diferentes receptores nucleares seleccionan, incluidos los PPAR, con la finalidad de ensamblar la maquinaria transcripcional al promotor del gen diana correspondiente30. Por tanto, hay una competición clara entre los diversos receptores nucleares. Las diferencias tanto en la expresión de estos factores y cofactores, como en la interacción de una o varias de estas proteínas tras la adición de los ligandos, podrían modular la capacidad del PPARα para regular la expresión de los genes diana. Esto podría explicar las diferencias encontradas en la respuesta a agonistas entre especies o entre diversos tipos de célulasbb0275, bb0370. No obstante, ratones carentes de PPARα, que expresan en sus hígados PPARα humano, responden funcionalmente a Wy-14,643 y a fenofibrato de manera similar a ratones que sí expresan PPARα (wild-type), controlando la expresión de genes diana conocidos75. Sin embargo, los ratones que expresan PPARα humano no muestran proliferación peroxisómica ni hepatocarcinoma, a diferencia de lo que se observa en ratones que presentan PPARα de ratón, a pesar de que los valores de expresión del PPARα sean similares en los 2 tipos de ratones75. Bien es verdad que el PPARα humano y el de ratón no son exactamente idénticos en sus dominios de unión al ADN y al ligando76, y se ha visto que el cambio de un solo aminoácido en el LBD del PPARα altera profundamente su actividad transcripcional77.
Parece ser que también hay una competición entre los diferentes isotipos de PPAR78. El subtipo PPARβ se ha propuesto que actúa como antagonista fisiológico del PPARα en humanos78, por lo que la mayor expresión del PPARβ en células humanas que en células de roedores podría implicar una reducción de la actividad del PPARα en células humanas55. Sin embargo, otros autores han propuesto que las funciones de los PPAR α y β pueden ser redundantes como reguladores transcripcionales, por lo que se ha indicado que el subtipo beta podría compensar la deficiencia del PPARα79. De una forma similar, la función del PPAR podría antagonizarse mediante la dimerización del receptor hepático X de tipo alfa (liver X receptor alpha, LXRα) con el PPARα o con el RXRα, lo que genera un complejo incapaz de unirse a su elemento respuesta en el ADN46. Para complicar aún más el asunto, mientras que ciertos ácidos grasos pueden actuar tanto de agonistas de PPARα, como de antagonistas del LXR, los ésteres de fibratos pueden presentar actividad tanto agonista como antagonista con el LXR80.
Aún más, el complejo PPAR:RXR también reconoce el elemento respuesta a estrógenos (ERE)81. Por ello, hay la posibilidad de una reacción hormonal cruzada a través del ERE81 y que algunos genes puedan estar regulados de una manera totalmente opuesta por el receptor de estrógenos (ER) y por el heterodímero PPAR:RXR. Así, por ejemplo, estudios llevados a cabo en ratones normales (wild-type), en roedores deficientes en receptor LDL, y en ratones sometidos a una ovariotectomía, han indicado el papel fundamental de los estrógenos en la regulación por fibratos de la obesidad y de la hipertriacilglicerolemia82. De hecho, en un estudio reciente realizado con ratones ovariotectomizados, el tratamiento concomitante con fenofibrato y estradiol mostró que esta hormona revierte los efectos del fibrato en los lípidos plasmáticos, así como en la expresión hepática de los genes diana del PPARα83. De forma similar, en ratas gestantes a término tratadas con fenofibrato, podría haber una competición entre el fibrato y el estradiol, cuyos valores son extremadamente altos al final de la gestación84, por ser la molécula que lidere el metabolismo lipídicobb0190, bb0425. Además, y de acuerdo con todo esto, parece ser que el sexo del animal influye en cómo responde a cambios inducidos en la expresión de PPARα, ya que se han encontrado diferencias relacionadas con el sexo del animal en los fenotipos resultantes de ratones carentes de PPARα o en los resultados obtenidos tras la utilización de ARN pequeño de interferencia (siRNA, del inglés small interfering RNA) para anular la expresión de PPARα86. Curiosamente, la atorvastatina, un agente hipolipemiante que como otras estatinas es capaz de inducir la expresión hepática de PPARα en ratasbb0435, bb0440, produce una respuesta muy dispar entre ratas viejas macho y hembra. De hecho, las ratas viejas hembras son prácticamente resistentes a los efectos de la estatina en el metabolismo lipídico, y ello se achaca a que probablemente los valores altos de estrógenos están inhibiendo la actividad transcripcional del PPARα89. Sin embargo, no puede descartarse la posibilidad de que los proliferadores peroxisómicos puedan también producir cambios en los valores séricos de estradiol85. De hecho, se ha visto que los proliferadores peroxisómicos pueden alterar la expresión de algunos genes relacionados con la síntesis de hormonas esteroi-deas90, aunque, hasta la fecha, sólo se ha encontrado uno de estos genes que esté regulado de manera dependiente por el PPARαbb0425, bb0450.
Fosforilación y otras modificaciones postraduccionalesEl proceso de fosforilación-desfosforilación es un importante mecanismo de regulación de la actividad transcripcional de los receptores nucleares. De hecho, el PPARα es una fosfoproteína y su función se encuentra influida por la acción de quinasas y fosfatasas92. En la región N-terminal, cercano al DBD del PPARα, se encuentra el dominio AF-1 que puede fosforilarse93, por lo que la actividad del PPARα depende de su estado de fosforilación94. De esta forma, es posible que la activación del PPARα por mecanismos independientes del ligando esté asociada con procesos dependientes de quinasas92. Los activadores de la proteína quinasa A (PKA) pueden aumentar la actividad del PPARα, tanto en ausencia como en presencia de agonistas de este receptor95. No obstante, se ha observado que la fosforilación del PPARα puede estar aumentada por tratamiento con proliferadores peroxisómicos, como ciprofibrato40. Además, el tratamiento de células con inhibidores de fosfatasas disminuye los valores de expresión de los genes inducidos por el ciprofibrato94. La insulina también aumenta la fosforilación del PPARα, e incrementa su actividad transcripcional96. Por otro lado, se ha visto que la actividad del PPARα también es modulada por la proteína quinasa activada por mitógenos (MAPK)93. De esta forma, la inhibición de la MAPK reduce la actividad del PPARα92. Además, inhibidores de la enzima HMG-CoA reductasa, como la cerivastatina, que actúan a través de la isoprenilación de la proteína G pequeña Rho A, pueden regular la señal de MAPK y, con ello, modular la actividad transcripcional del PPARα97. Ésta es una interferencia (cross-talking) importante entre los fibratos y las estatinas92. A su vez, la modificación del estado de fosforilación del PPAR afecta a su afinidad por el ligando46.
Además de la fosforilación de la proteína, la S-nitrosilación puede ser crítica en la regulación por receptores nucleares de las funciones celulares vitales. El óxido nítrico (NO) interacciona con residuos de cisteína formando un enlace nitrosotiol, que altera la actividad de este tipo de proteínas. Así, por ejemplo, el NO inhibe significativamente la unión del ER al ADN a través de su ERE. Es más, el ER contiene una zona con 2 dedos de cinc y 4 cisteínas en cada uno, con una elevada homología con otros miembros de la superfamilia de receptores nucleares, como los PPAR98.
Se han identificado numerosas proteínas similares a la ubiquitina, capaces de unirse a residuos de lisina en proteínas diana. La más conocida es el pequeño modificador relacionado con la ubiquitina (SUMO, del inglés small ubiquitin-related modifier) que agrupa una serie de péptidos de unos 100 aminoácidos cada uno, que son capaces de unirse covalentemente a la proteína diana a través de residuos específicos de lisina. La sumoilación de las proteínas tiene funciones variadas, como, por ejemplo, regular la estabilidad de la proteína, su localización nuclear, así como la transcripción génica99. La sumoilación de la lisina en posición 107 del PPARγ disminuye su activación transcripcio-nal100. La posibilidad más factible es que la sumoilación modifique localmente o totalmente la conformación del dominio AF-1, donde se encuentra un sitio de sumoilación, y modifique la capacidad del PPARγ para interaccionar con sus correguladores. El PPARα también presenta sitios potenciales de sumoilación en la región D. Sin embargo, parece ser que la sumoilación es una modificación más específica del PPARγ que del resto de subtipos de PPAR101.
Funciones del PPARαEfectos en el metabolismo lipídicoEfectos en el catabolismo de las VLDLLa LPL es una enzima clave en el metabolismo de las lipoproteínas que hidroliza los triacilglicéridos (TG) de los quilomicrones y las VLDL102. Estudios in vivo indican que, mientras los fibratos y los ácidos grasos inducen claramente un aumento en los valores de ARNm de LPL en el hígado (Tabla 1), se han encontrado resultados contradictorios tanto en el tejido adiposo, como en otros tejidos extrahepáticosbb0145, bb0345, bb0515. Por lo tanto, el efecto de los agonistas del PPARα en la eliminación del plasma de las lipoproteínas ricas en TG parece deberse principalmente a cambios en la expresión hepática de los inhibidores (apo C-III) y activadores (apo A-V) de la actividad de la LPL (Tabla 1)29. De hecho, los valores de apo C-III parecen estar correlacionados positivamente con las concentraciones plasmáticas de TG. Se han indicado diversos mecanismos para explicar los efectos de la activación del PPARα en la regulación de la expresión del gen de la apo C-III: el primero, sería una competición entre PPAR y HNF-4 por la unión al promotor de la apo C-III. La sustitución de un activador fuerte como el HNF-4, por otro menos activo, como el complejo PPAR:RXR, explicaría una reducción en la expresión de la apo C-III, la cual dependería, por tanto, de la abundancia relativa en la célula de PPARα y HNF-4. No obstante, se ha observado también que los agonistas del PPARα disminuyen los valores de proteína del HNF-433. El segundo mecanismo de represión de la apo C-III por los fibratos podría ocurrir por un aumento dependiente del PPARα en la expresión de un regulador negativo de la transcripción. De acuerdo con esto, los activadores del PPARα inducen la expresión hepática de Rev-erbα, un receptor nuclear huérfano que es un potente represor de la transcripciónbb0520, bb0525. A su vez, el PPARα puede reprimir la transcripción de la apo C-III al interaccionar físicamente con otros factores de transcripción, lo que conlleva la formación de complejos inactivos y, por tanto, limita la inducción de la expresión de la apo C-III. Este parece ser el caso del factor transcripcional positivo de la familia forkhead (FoxO1, del inglés forkhead box o1)106. Por otro lado, recientemente se ha descubierto que la apo A-V participa en la regulación de los valores plasmáticos de TG en humanos y roedores, siendo el primer gen que codifica para una apolipoproteína, cuyo aumento en la expresión produce una disminución de los valores de TG. De acuerdo con esto, hepatocitos primarios humanos tratados con Wy-14,643 o fenofibrato mostraron una inducción fuerte de los valores del ARNm que codifica para apo A-V73, a través de la activación del PPARα y su unión al PPRE funcional. Estos estudios demuestran que la apo A-V es uno de los genes humanos que responde de forma más eficaz a la acción del PPARα (Tabla 1, Fig. 1)73. No obstante, no se ha identificado aún un PPRE en el promotor de la apo AV en la rata, lo que indicaría probablemente otra diferencia adicional entre especies107. En resumen, estos hechos muestran que los activadores del PPARα aumentan el catabolismo de las VLDL, lo que significa cambios en la composición de las lipoproteínas de baja densidad (LDL) y, con ello, un aumento en su captación del plasma por los tejidos.
Tabla 1. Principales rutas del metabolismo lipídico y genes diana regulados por el receptor activado por proliferadores peroxisómicos
| Genes diana | Función del gen | PPRE |
| Transporte extracelular | ||
| Apo A-I | Transporte de lípidos por la sangre | Sí |
| Apo A-II | Transporte de lípidos por la sangre | Sí |
| Apo A-V | Transporte de lípidos por la sangre | Sí |
| Apo C-III | Transporte de lípidos por la sangre | No |
| LPL | Liberación de ácidos grasos de TG de lipoproteínas | Sí |
| SR-B1 | Receptor de HDL | Sí |
| Transporte intracelular | ||
| FATP | Transporte de ácidos grasos a través de la membrana celular | Sí |
| FAT/CD36 | Transporte de ácidos grasos a través de la membrana celular | Sí |
| FABP | Unión de ácidos grasos en el ámbito intracelular | Sí |
| ACBP | Unión de acil-CoA en el ámbito intracelular | Sí |
| Activación de ácidos grasos | ||
| ACS | Activación de ácidos grasos | Sí |
| Lipogenia | ||
| EM | Producción de NADPH | Sí |
| ACC | Síntesis de ácidos grasos | No |
| FAS | Síntesis de ácidos grasos | No |
| SCD-1 | Desaturación de acil-CoA | Sí |
| Oxidación | ||
| Mitocondrial | ||
| CPT-1 | Entrada de acil-CoA | Sí |
| MCA | β-oxidación | Sí |
| Peroxisomal | ||
| ACO | β-oxidación | Sí |
| HD (L-PBE) | β-oxidación | Sí |
| TIO | β-oxidación | No |
| D-PBE (Hsd17b4) | β-oxidación | No |
| Microsomal | ||
| CYP4A1 | ω−oxidación | Sí |
| CYP4A6 | ω−oxidación | Sí |
| Cetogenia | ||
| HMG-CoA sintasa | Síntesis de cuerpos cetónicos | Sí |
| Termogenia | ||
| UCP-2 | Termogenia | Sí |
PPARα está implicado en la regulación del transporte intracelular y extracelular de ácidos grasos, la activación de éstos, la lipogenia, la oxidación mitocondrial, peroxisomal y microsomal, la síntesis de cuerpos cetónicos y la termogenia. Se indican, además, los genes para los que se ha descrito un elemento respuesta a proliferadores peroxisómicos (PPRE) funcional.
ACBP: proteína de unión de acil-CoA; ACC: acetil-CoA carboxilasa; ACO: acil-CoA oxidasa; ACS: acil-CoA sintetasa; Apo: apolipoproteína; CPT-1: carnitina palmitoil transferasa 1; EM: enzima málica; FABP: proteína de unión a los ácidos grasos; FAS: ácido graso sintasa; FAT: transportador de ácidos grasos; FATP: proteína transportadora de ácidos grasos; HD: hidratasa deshidrogenasa; HDL: lipoproteínas de alta densidad; Hsd17b4: proteína D-bifuncional; LPL: lipoproteína lipasa; MCAD: acil-CoA deshidrogenasa de cadena media; nAdPH: nicotinamida-adenina dinucleótido fosfato; SCD-1: estearoil-CoA desaturasa 1; SR-B1: receptor scavenger de tipo B1; TG: triacilglicéridos; TIO: 3-cetoacil-CoA tiolasa; UCP: proteínas desacoplantes.
Todo proceso que module la captación y la metabolización de ácidos grasos por el hígado para su conversión en TG afectará la producción de VLDL. El PPARα desempeña un papel importante en el metabolismo intracelular de los ácidos grasos, ya que regula la expresión de genes implicados en diversos pasos cruciales del metabolismo lipídico. Así, el PPARα puede regular la secreción de VLDL mediante distintos mecanismos. El primero de todos, la captación celular de ácidos grasos de cadena larga, se encuentra facilitado y regulado por transportadores de ácidos grasos, como la proteína transportadora de ácidos grasos (FATP) y el transportador de ácidos grasos (FAT), también llamado CD36. La expresión de ambos transportadores es aumentada en el hígado de rata por fibratos y por ácidos grasos (Tabla 1, Fig. 1)102. Mientras que los transportadores de membrana facilitan el paso de los FFA a través de la membrana plasmática, la acil-CoA sintetasa (ACS) cataliza la esterificación de estos FFA con la coenzima A y genera derivados acil-CoA, lo que previene su salida de la célula, y activa esos ácidos grasos para su utilización tanto en rutas catabólicas como anabólicas. La expresión y la actividad del gen ACS son inducidas por fibratos y ácidos grasos en una gran variedad de tejidos y células (Tabla 1, Fig. 1)108. Una vez en el citoplasma, los FFA se unen a proteínas transportadoras, como la proteína de unión de ácidos grasos (FABP) y la proteína de unión de acil-CoA (ACBP). Isseman y Green109 identificaron en roedores un PPRE en el promotor del gen que codifica para la FABP. Así, los fármacos hipolipemiantes son capaces de inducir la expresión de FABP vía PPARα (Tabla 1, Fig. 1)110. Por otro lado, algunos estudios han indicado que el complejo PPAR:RXR aumenta la expresión de FABP por competición con COUP-TFII por el sitio DR-1 en el promotor proximal del gen. De esta forma, el contenido relativo celular de COUP-TFII (represor) y PPAR:RXR (activador) determina la expresión diferencial de FABP que se ha encontrado en diversos tipos celulares111.
Un proceso que también puede limitar la disponibilidad de ácidos grasos para su conversión en TG y posterior formación de VLDL es el catabolismo de ácidos grasos29. El papel crítico de los agonistas del PPARα en la regulación de la oxidación de los ácidos grasos ha sido bien documentado. De acuerdo con esto, los valores más elevados de expresión del PPARα se observan en tejidos con un activo catabolismo de ácidos grasos. Como ya se ha comentado, la proliferación peroxisómica fue uno de los primeros efectos descritos en roedores tras la exposición a activadores sintéticos del PPARα y es un proceso que se ha relacionado con un aumento en la β-oxidación peroxisómica de los ácidos grasos112. Esta ruta enzimática es la causa del metabolismo de los ácidos grasos de cadena larga en la que están implicadas secuencialmente las enzimas ACO, enoil-CoA hidratasa/3-hidroxiacil-CoA deshidrogenasa (enzima L-bifuncional, HD) y 3-cetoacil-CoA tiolasa (TIO)113. Los intermediarios L- y D-hidroxi que se generan en el primer paso de la β-oxidación, posteriormente pueden metabolizarse tanto por la enzima HD, como por la proteína D-bifuncional (Hsd17b4)114. Las actividades de todas estas enzimas son estimuladas en respuesta a agonistas PPARα, lo cual se debe a un aumento en la tasa de transcripción de sus genes (Tabla 1, Fig. 1)115. Así, por ejemplo, Corton et al91 mostraron que la expresión de Hsd17b4 aumentaba significativamente tras el tratamiento con proliferadores peroxisómicos a roedores. Más aún, la expresión de la proteasa Tysnd1 (trypsin domain containing 1) es inducida por el bezafibrato, un agonista del PPARα. Recientemente, Kurochkin et al116 han propuesto un modelo interesante que indica que Tysnd1 interviene en el procesamiento de las enzimas peroxisómicas, ACO, Hsd17b4 y TIO, lo que permite su ensamblaje formando un complejo supramolecular con el objetivo de incrementar aún más la tasa de la β-oxidación.
El análisis del promotor del gen de la ACO condujo a la identificación de 2 PPRE que median en la activación transcripcional por el PPARα10. Poco después, también se caracterizó un PPRE en la secuencia reguladora de la HD117. Sin embargo, aunque la TIO es también inducida transcripcionalmente por proliferadores peroxisómicos, hasta el momento no se ha identificado un PPRE funcional en este gen102. No obstante, el metabolismo de los ácidos grasos se regula principalmente en el ámbito de la β-oxidación mitocondrial de los ácidos grasos. El primer paso limitante en esta β-oxidación es la entrada de los ácidos grasos a la mitocondria, que se encuentra controlada por un sistema transportador dependiente de carnitina. La carnitina palmitoil transferasa 1 (CPT-1) cataliza la formación de acilcarnitina para su translocación a través de la membrana mitocondrial interna. La expresión del gen que codifica para la CPT-1 es aumentada por ácidos grasos y por proliferadores perosixómicos (Tabla 1, Fig. 1)112, y se ha caracterizado además un PPRE funcional en el promotor del gen de la CPT-1118. No obstante, en hepatocitos se ha observado que los inhibidores de la lipooxigenasa impiden la activación por fibratos de la expresión de CPT-1, si bien no afectan a la inducción por ácidos grasos119. En ese mismo sentido, los proliferadores peroxisómicos, pero no los ácidos grasos de cadena larga, inducen la CPT-2 en hepatocitos fetales de rata120. Además, el PPARα regula a otro nivel la β-oxidación mitocondrial, al modular también la expresión del gen de la acil-CoA deshidrogenasa de cadena media (MCAD) (Tabla 1, Fig. 1)121. Uno de los destinos de las unidades de acetil-CoA producidos por la β-oxidación de los ácidos grasos en la mitocondria es convertirse en cuerpos cetónicos, principalmente acetoacetato y 3-hidroxibutirato. La hidroximetilglutaril-CoA (HMG-CoA) sintasa mitocondrial es la principal enzima involucrada en la formación de cuerpos cetónicos y se encuentra directamente controlada por el PPARα (Tabla 1, Fig. 1)122. Por otro lado, la β-oxidacion mitocondrial contribuye enormemente en la producción de energía mediante la fosforilación oxidativa, generando ATP. Sin embargo, el PPARα parece aumentar el gasto energético al inducir la expresión de proteínas desacoplantes (UCP)123, como UCP1, UCP2 (Tabla 1) y UCP3, las cuales son transportadores mitocondriales de la membrana mitocondrial interna que actúan disipando el gradiente de protones y aumentando la termogenia, al tiempo que reducen la eficacia en la síntesis de ATP124. Por otro lado, el sistema citocromo-monooxigenasa desempeña un papel central en la oxidación de una amplia variedad de compuestos, tanto endógenos como exógenos. Las enzimas CYP4A microsomales participan en el sistema como un grupo aparte de la superfamilia del citocromo P450. Estas enzimas catalizan la ω-hidroxilación de ácidos grasos y eicosanoides, como el leucotrieno LTB4. De hecho, la ω-hidroxilación es el primer paso en la neutralización del LTB4, para luego degradarse completamente a través de la β-oxidación en los peroxisomas112. Al menos 2 genes de la familia CYP4A, CYP4A1 y CYP4A6 contienen un PPRE funcional en su promotor y, por tanto, responden in vivo y en cultivos celulares a activadores del PPAR (Tabla 1)bb0625, bb0630.
El PPARα también está involucrado en la síntesis de ácidos grasos. Se ha visto que el gen de la enzima málica (EM) está regulado positivamente por proliferadores peroxisómicos vía PPARα127, a través de un PPRE (Tabla 1, Fig. 1). La reacción catalizada por la EM es una descarboxilación oxidativa del malato citosólico, lo que genera piruvato y conduce a la formación de nicotinamida-adenina dinucleótido fosfato (NADPH), necesario para la síntesis de lípidos. Sin embargo, el papel del PPARα parece complejo, ya que otros genes lipogénicos importantes, como la acetil-CoA carboxilasa (ACC) y la ácido graso sintasa (FAS)128 son regulados negativamente por los PufA128 y por proliferadores peroxisómicos (Tabla 1)129. Además, la enzima es-tearoil-CoA desaturasa 1 (SCD-1), que cataliza la Δ9-desaturación de los ácidos grasos saturados, se regula positivamente mediante fibratos, pero negativamente mediante PUFA (Tabla 1)130. En cuanto a la expresión de genes relacionados más directamente con la síntesis de TG, se ha identificado recientemente al PPARα como un estimulador del gen que codifica para la diacilglicerol aciltransferasa (DGAt)29. No obstante, se ha demostrado que los agonistas del PPARα aumentan la actividad de DGAT en el citoplasma y la disminuyen en el retículo endoplásmico, lo que parece ser la causa de que los TG se acumulen en gotas grasas en el citoplasma, en vez de incorporarse en las VLDLbb0145, bb0655.
Efectos en el catabolismo de las lipoproteínas de alta densidadEl papel principal de las lipoproteínas de alta densidad (HDL) se encuentra en la eliminación del exceso de colesterol y su transporte desde los tejidos periféricos hacia el hígado, en el denominado transporte reverso de colesterol, y su efecto protector contra el desarrollo de la enfermedad coronaria. El PPARα parece ser la principal isoforma implicada en el metabolismo de las HDL29. Las apo A-I y apo A-II son los principales constituyentes proteicos de la HDL. Es interesante señalar que los activadores del PPARα afectan al metabolismo de las HDL de manera opuesta en roedores y en humanos. Mientras que el tratamiento con fibratos a ratas disminuye el valor plasmático de HDL, en humanos, por lo general, se observa un aumentobb0660, bb0665. Este aumento en los valores plasmáticos de HDL está relacionado, al menos en parte, con cambios en la expresión hepática de apo A-I y apo A-II. Los activadores del PPARα provocan en humanos un aumento en la transcripción de los genes que codifican para apo A-I y apo A-II (Tabla 1, Fig. 1)bb0670, bb0675. Por el contrario, estos genes se reprimen en roedores136. Vu-Dac et al135 observaron que el PPARα se une al PPRE localizado en el promotor del gen de la apo A-I humana. Sin embargo, en roedores, la regulación negativa por fibratos de la expresión del gen de la apo A-I parece ser más compleja. La falta de inducción transcripcional de la apo A-I de rata se debe a una diferencia de 3 nucleótidos entre los promotores de rata y de humano, lo que impide la unión del PPARα al PPRE de roedores105. Por el contrario, el Rev-erbα sí que puede unirse a un elemento respuesta negativo cercano a la caja TATA, el cual está presente sólo en el promotor de la apo A-I de rata105. Dado que los fibratos inducen la expresión de Rev-erbα la expresión de apo A-I en roedores puede reprimirse mediante este mecanismo.
Por ende, los fibratos también afectan a la expresión de enzimas y receptores que regulan el metabolismo de las HDL. En rata, los fibratos disminuyen la producción de las enzimas que modifican las HDL, como la lipasa hepática y la lecitina colesterol aciltransferasa (LCAT)bb0685, bb0690. Además, los agonistas del PPAR regulan la expresión de los receptores de HDL. En ratón, el receptor scavenger de tipo B1 (SR-B1), y su homólogo humano CLA-I, se han identificado como receptores de HDL, los cuales unen HDL con alta afinidad, median la captación selectiva de ésteres de colesterol por el hígado y por tejidos esteroidogénicosbb0695, bb0700, y promueven además la eliminación de colesterol de los tejidos periféricos. De hecho, el tratamiento de macrófagos humanos con activadores del PPARα provocó una inducción en la expresión de CLA-I, sugiriendo que el PPARα promueve en macrófagos la salida de colesterol hacia las HDL29. Además, se ha encontrado que el SR-B1 es inducido en aortas de ratones deficientes en apo E después del tratamiento con agonistas del PPAR (Tabla 1, Fig. 1). Por el contrario, los fibratos, a través del PPARα, reducen la expresión del gen que codifica para el SRB1 en el hígado de roedores141.
Efectos en el metabolismo de la glucosaLos PPAR también desempeñan un papel importante en la homeostasis glucídica. Los agonistas del PPARα, a través de un aumento en la oxidación de los ácidos grasos y en la producción de los cuerpos cetónicos, promueven un ahorro en el gasto de glucosa. De acuerdo con esto, ratones carentes de PPARα presentan hipoglucemia durante el ayuno, debida a una menor capacidad gluconeogénica hepática y a una disminución en la capacidad de oxidar ácidos grasos142. Además, algunos estudios indican un efecto beneficioso de la activación del PPARα en la sensibilidad insulínicabb0715, bb0720. De este modo, el tratamiento con fibratos disminuye de forma eficaz la hiperinsulinemia y la hiperglucemia observadas en ratones alimentados con una dieta rica en grasas o en ratones genéticamente resistentes a la insulinabb0715, bb0725.
De una manera más específica, la piruvato cinasa hepática (L-PK) es una enzima glucolítica que desempeña un papel clave en el metabolismo glucídico y lipídico. La transcripción del gen de la L-PK es inhibida por PUFA ω-3 y por activadores del PPARα. Además, los PUFA y Wy-14,643 interfieren, aunque por mecanismos diferentes, en la estimulación por glucosa de la transcripción del gen de la piruvato cinasa, tanto in vivo como en hepatocitos primarios de rata146. Por otro lado, la piruvato deshidrogenasa cataliza la transformación de piruvato en acetil-CoA y, por tanto, controla la transformación del piruvato en diversos compuestos metabólicos, como ácidos grasos, aminoácidos o su oxidación completa. El PPARα también ejerce un papel directo en la regulación de la gluconeogenia mediante la estimulación de la expresión de la piruvato deshidrogenasa quinasa 4 (PDK4)147. Esta enzima inactiva, mediante fosforilación, el complejo piruvato deshidrogenasa, y promueve la utilización de piruvato en la gluconeogenia más que su oxidación a acetil-CoA. En ratones carentes de PPARα ayunados, la síntesis de glucosa desde lactato se encuentra fuertemente reducida, lo que demuestra que el PPARα también influye en la utilización de este sustrato para la producción de glucosa en el hígado. Sin embargo, la producción hepática de glucosa en ayunas fue sorprendentemente mayor en ratones carentes de PPARα que en los animales normales, debido a una aumentada producción de glucosa desde glicerol en esos animales148. Estos datos revelan que la hipoglucemia grave observada en ratones carentes de PPARα durante el ayuno no se debe a una menor producción de glucosa en el hígado, sino a un aclaramiento mayor de glucosa a otros tejidos, como por ejemplo una captación mayor de glucosa por el tejido adiposo149. Otra enzima de la ruta de biosíntesis de glucosa que se encuentra regulada por el PPARα es la PEPCK, que cataliza un paso limitante de la gluconeogenia. No obstante, hay resultados contradictorios respecto a la consideración de la PEPCK como gen diana del PPARα en el hígado45, dado que la expresión del gen que codifica para la PEPCK está estimulada en estado de ayuno, tanto en ratones normales como en ratones carentes del PPARα142. De hecho, aunque el promotor del gen de la PEPCK contiene 2 secuencias DR1150, este gen sólo es sensible a la acción de los ácidos grasos en adipocitos, pero no en hepatocitos70.
Efectos en el metabolismo proteicoLos aminoácidos, además de ser sustratos para la síntesis de neurotransmisores, nucleótidos y poliaminas, también desempeñan un papel importante en el aporte de energía. El mantenimiento del equilibrio nitrogenado es determinado por la homeostasia de la glutamina y el ciclo de la urea19. El PPARα actúa como un regulador negativo del ciclo de la urea, e inhibe la expresión de enzimas de este ciclo, como la carbamoil fosfato sintasa tipo I (CPS-I), entre otras151. Las 2 fuentes principales de la producción de amonio en el organismo son el catabolismo de las proteínas de la dieta y la degradación de proteínas endógenas que ocurre después de un ayuno prolongado. Mientras que ratones normales tras el ayuno presentan una disminución en los valores de urea en plasma, en ratones carentes de PPARα permanecen elevados. Puesto que el HNF-4 y el C/EBPα son reguladores positivos del ciclo de la urea, el modo de regulación de esta vía podría ser el resultado del equilibrio entre la activación (por HNF-4 y/o C/EBP) y la inhibición (por PPARα) de la actividad de este ciclobb0740, bb0755.
Efectos en la inflamación vascular y hepáticaEl papel de la inflamación en la aterosclerosis está bien establecido18. Las lesiones vasculares de la aterosclerosis no son sólo el resultado de la acumulación de lípidos, sino también el resultado de un daño vascular que conduce a la activación de una respuesta específica celular y molecular, tanto en la pared vascular como en el sistema inmunitario152. En relación con esto, los PPAR se expresan en células del sistema vascular y del sistema inmunitario: monocitos, macrófagos, linfocitos, EC y VSMC30. Se ha demostrado claramente el papel del PPARα en la inflamación en ratones carentes de PPARα, los cuales presentan una respuesta inflamatoria prolongada tras la estimulación provocada por leucotrienos y ácido araquidónico27. La activación del PPARα por el LTB4 induce la β-oxidación peroxisómica de estos leucotrienos en el hígado de roedores, lo que conlleva un control negativo retroinhibitorio del estímulo inflamatorio provocado por este lípido que no se da en los ratones carentes de PPARα46. No obstante, otros autores153 han cuestionado esta regulación negativa del metabolismo de los LTB4 en hepatocitos mediada por el PPARα. Los PPAR controlan diversos mecanismos en el ámbito vascular que incluyen: la selección y la activación de células, la respuesta inflamatoria local, la constricción vascular, y la migración celular y la trombosisbb0090, bb0240. Como ya se ha comentado anteriormente, el PPARα inhibe la expresión de genes marcadores de inflamación inducidos por NF-κB, como la molécula de adhesión celular vascular 1 (VCAM-1), la ciclooxigenasa 2 (COX-2) y las interleucinas (IL-6 e IL-1) (Fig. 1), lo que daría una explicación molecular a los efectos antiinflamatorios de los ligandos del PPARα in vivo. Es más, de forma similar a los agonistas sintéticos de PPARα, las VLDL tratadas con LPL reprimen la expresión de VCAM-1 inducida por citocinas en EC de ratones normales (wild-type), pero no en las células de ratones carentes de PPARα58. Recientemente se han descrito los mecanismos moleculares que están implicados en la interacción del PPARα con la señal inflamatoria a diferentes niveles152. El PPARα también inhibe la producción de endotelina 1 (ET-1) y factor tisular (TF) en EC, VSMC y macrófagos (Fig. 1)bb0770, bb0775, y aumenta la expresión de la sintasa endotelial de óxido nítrico (eNOS) en células vasculares156. Todos estos efectos mejoran la función endotelial, aumentan la producción de óxido nítrico156, y generan un efecto vasodilatador protector. En células humanas de la musculatura lisa aórtica, la activación del PPARα por fibratos inhibe la secreción de IL-6 inducida por iL-1157. Además, Delerive et al31 han observado que las aortas de ratones carentes de PPARα presentan una respuesta inflamatoria exagerada tras la estimulación por lipopolisacáridos (LPS), y demuestran in vivo que la actividad antiinflamatoria de los agonistas del PPARα requiere la expresión del receptor nuclear. La metaloproteinasa de la matriz 9 (MMP-9)158 y el TF159 son proteínas localizadas en el macrófago que promueven la inestabilidad de la lesión y la coagulación, respectivamente. Los agonistas del PPARα inhiben in vitro la expresión de estas proteínas en macrófagos (Fig. 1), y aportan un posible mecanismo por el cual los agonistas del PPARα estabilizan ateromas y reducen la trombogenia.
Dado que el PPARα se expresa en valores muy elevados en hígado, era de esperar que tuviera un papel regulador de la respuesta inflamatoria en el hígado152. La respuesta de fase aguda es un proceso inflamatorio clave para que se inicien los mecanismos de defensa. Sin embargo, puede ocasionar alteraciones importantes si la activación se hace crónica. Tanto la IL-6 como la IL-1 estimulan la producción de proteínas de fase aguda, como la proteína C reactiva (PCR), el fibrinógeno y el amiloide sérico A (SAA), los cuales son marcadores de enfermedad cardiovascular. Los fibratos actúan como reguladores negativos de todas estas y otras proteínas de fase aguda positivas (Fig. 1)bb0800, bb0805. Se ha indicado que la base molecular de la inhibición de la expresión inducida por IL-6 de las cadenas del fibrinógeno α y β y de la expresión de SAA, se debe a la interferencia del PPARα con la señal del C/EBP34. Se ha descrito un mecanismo similar, pero que interfiere con la señal del NF-κB, para la inhibición mediada por fibratos de la expresión de PCR inducida por IL-1 (Fig. 1)162. Por tanto, la expresión de proteínas de fase aguda está regulada negativamente por activadores del PPARα, y sus valores en plasma se encuentran disminuidos en humanos después del tratamiento con fibratos. De hecho, el tratamiento con fenofibrato en pacientes dislipémicos disminuye las concentraciones en plasma de fibrinógeno, SAA, IL-6 y PCR163, mientras que los valores de albúmina, una proteína de fase aguda negativa, aumentan152.
Efectos inesperados de la activación del PPARαDesde hace décadas, el uso de los fibratos como agentes hipolipemiantes ha demostrado su seguridad y eficacia para disminuir la lipemia y como antiinflamatorios, parámetros fundamentales en la prevención de las enfermedades cardiovasculares. La activación de PPARα se ha comprobado que reduce los síntomas del síndrome metabólico y otras manifestaciones de resistencia a la insulina, como son obesidad visceral, resistencia a la insulina, inflamación y dislipemia aterogénica, o lo que es lo mismo, valores bajos de HDL, triacilglicerolemia elevada, y LDL pequeñas y densas. Sin embargo, diversos trabajos han revelado que esos agonistas del PPARα pueden potencialmente generar efectos adversos o presentar una capacidad hipolipemiante disminuida, así como una acción antiinflamatoria ineficiente.
De cualquier manera, bien es verdad que los diversos factores que pueden modular la actividad del PPARα (su contenido celular, la naturaleza del ligando, las reacciones hormonales cruzadas, la estructura del PPRE e incluso las modificaciones postraduccionales del receptor) podrían explicar, al menos en parte, por qué los fármacos activadores de PPARα muestran en ocasiones una acción hipolipemiante tan variable, a veces incluso ineficiente, o por qué se obtienen resultados tan diversos en estudios clínicos cardiovasculares en los que se han usado estos agentes164, o bien por qué producen efectos secundarios58, que en algunos casos han limitado su uso.
Metabolismo lipídicoAunque hay diversos estudios que muestran una relación inversa entre el contenido hepático del ARNm que codifica para el PPARα y los valores plasmáticos de TGbb0440, bb0825, otros estudios han descrito que los fibratos pueden fallar o corregir de manera insuficiente la hipertriacilglicerolemia en determinadas condiciones. Entre ellos, un tratamiento agudo con fenofibrato a ratas Zucker hipertriacilglicerolémicas y obesas provocó un aumento marcado en los TG cuando se comparó con ratas controles obesas no tratadas166. En relación con esto, en otro estudio se observó que las ratas Zucker no tratadas presentaban un aumento en los TG plasmáticos después de 4 semanas, cuando se comparaban con el día inicial del experimento. Es de reseñar que, en este mismo estudio, el tratamiento semicrónico con fenofibrato solamente fue capaz de evitar ese aumento en los valores de TG observado en las ratas controles, mientras que la rosiglitazona, un ligando del PPARγ, sí era capaz de reducir de forma eficaz la hipertriacilglicerolemia167. Bien es verdad que, en paralelo con las ratas Zucker, se usaron ratones db/db, y en estos animales el fenofibrato administrado durante 2 semanas fue capaz de evitar, e incluso disminuir, la hipertriacilglicerolemia observada en los ratones control, de forma similar a cómo lo hacía la rosiglitazona167. En otro estudio, en el que se comparaban los efectos de los fibratos en ratones alimentados con diversas dietas hiperlipidémicas, se observó que en los animales que recibieron dieta estándar, tanto fenofibrato como gemfibrocilo, los valores de TG disminuían, mientras que en los animales que recibieron dieta hipercolesterolémica, tan sólo el tratamiento con fenofibrato conseguía disminuir los valores de TG plasmáticos168. Una situación similar se observó en el grupo de animales alimentados con una dieta hipertriacilglicerolémica, ya que el tratamiento con fenofibrato provocó una disminución de los valores de TG plasmáticos, aunque no de forma dependiente de la dosis, y el tratamiento con gemfibrocilo ni siquiera afectó a la hipertriacilglicerolemia168. Más aún, en algunos casos se han encontrado efectos contrapuestos de diferentes fibratos, tanto en ratas normales como en hiperlipidémicas. Según esto, en un estudio en el que se usaron bezafibrato y gemfibrocilo, ambos fármacos consiguieron disminuir de forma similar los TG plasmáticos en las ratas alimentadas tanto con dieta normal, como hipertriacilglicerolémica. Sin embargo, el tratamiento con gemfibrocilo disminuyó el colesterol unido a las LDL (cLDL), pero elevó el colesterol unido a las HDL (cHDL), mientras que el bezafibrato provocó efectos opuestos169. Además, la administración de estos compuestos originó cambios opuestos en las concentraciones de TG hepáticos en las ratas hipertriacilglicerolémicas169. Los fibratos también mostraron efectos inesperados con otros modelos animales, como fue el caso de ratas al final de la gestación, las cuales presentan de forma fisiológica hipertriacilglicerolemia cuando se comparan con ratas no gestantes69. Es más, como se ha comentado para el caso de las ratas Zucker obesas, las ratas gestantes también presentan una hipertriacilglicerolemia que aumenta con el tiempo. De esta forma, aunque los valores de TG plasmáticos disminuían en las ratas gestantes durante los primeros 2 días de tratamiento con el fenofibrato, el efecto desaparecía al tercer día, e incluso se observaba un incremento de los TG plasmáticos en el cuarto día. Por el contrario, en las ratas vírgenes, el fenofibrato disminuyó los TG plasmáticos durante todo el experimento. Por otro lado, se ha observado una falta de sensibilidad a los activadores del PPARα en ratas vírgenes, en comparación con madres lactantes170. Los fibratos inducen la expresión del ARNm que codifica para la UCP-3 en el músculo esquelético de ratas durante la lactancia, mientras que no se consigue una activación similar en las ratas vírgenes. No obstante, dado que los FFA también inducen la expresión de UCP-3 y que los valores de FFA en suero son mucho más elevados en ratas no preñadas que en las madres lactantes, la falta de efecto de los fibratos en las vírgenes podría achacarse a que la ruta ya estaba completamente activada170. Por otro lado, otra situación en la que se observan respuestas inesperadas de los agonistas del PPARα es el envejecimiento. Las ratas viejas presentan una reducción drástica en la expresión y actividad hepáticas del PPARα. De hecho, se hacen resistentes al tratamiento con gemfibrocilo o con bezafibrato, ya que estos fármacos no consiguen la típica acción hipotriacilglicerolemiante, ni inducen un aumento significativo de la expresión hepática de los genes diana del PPARα, ni siquiera en dosis en las que sí son efectivos en animales jóvenesbb0820, bb0825. En definitiva, estos estudios indican que el efecto de los fibratos en la triacilglicerolemia podría depender del animal empleado, del grado de hipertriacilglicerolemia o incluso de la cantidad de ácidos grasos que se movilicen hacia el hígadobb0345, bb0835.
Igualmente, se han encontrado efectos inesperados de los fibratos en ratones transgénicos y knockout. Así, los ratones carentes de apo E, un modelo de ratón que presenta características similares a la dislipemia y aterosclerosis humanas, mostraban valores elevados de TG. Paradójicamente, el tratamiento de estos animales con fenofibrato aumentó los valores de TG cuando se compararon con los controles171. De acuerdo con ello, en ratones carentes de apo E tratados con cibrofibrato, se ha observado un aumento considerable de las lesiones ateroscleróticas, si se comparan con el efecto en los animales no tratados172. Es más, en ratones transgénicos que portan apo A-II humana, los cuales comparten algunas características fenotípicas con los ratones carentes de PPARα, la activación del PPARα por fibratos no era capaz de corregir la hiperlipemia combinada que presentaban. De hecho, tras 2 semanas de tratamiento con fenofibrato, estos ratones transgénicos con apo A-II humana mostraban un aumento inesperado de los TG plasmáticos, debido principalmente a un catabolismo menor de las VLDL y a un peor funcionamiento de la señalización mediada por el PPARα173.
En humanos también se han descrito casos en los que los fibratos han mostrado ser ineficientes o incapaces de corregir de forma eficaz la hipertriacilglicerolemia en determinadas circunstanciasbb0870, bb0875, como por ejemplo, en la hiperlipemia grave combinada, o en el tratamiento combinado con tratamiento hormonal sustitutivo en mujeres obesas posmenopáusicas176. Es interesante señalar que la hipertriacilglicerolemia es un indicador potente de enfermedad coronaria177. Ello podría significar que muchos pacientes no estarían recibiendo un tratamiento apropiado para la dislipemia que acompaña generalmente a la enfermedad cardiovascular, a las situaciones de resistencia a la insulina y/o al síndrome metabólico. En este sentido, la monoterapia con fibratos no es generalmente capaz de normalizar el perfil lipídico que acompaña a la hiperlipemia combinada grave. Realmente, los fibratos son más activos en la hipertriacilglicerolemia, mientras que las estatinas son más activas en la hipercolesterolemia, que son los 2 componentes de esta hiperlipemia175. En un estudio realizado con mujeres con sobrepeso, posmenopausia e hipertriacilgliceridemia, se observó que la combinación de un agente hipolipemiante del tipo fibrato junto con el estradiol que se administraba como tratamiento hormonal sustitutivo durante 3 meses, no sólo no tenía efectos beneficiosos en los perfiles lipídicos o lipoproteicos, sino que incluso producía un incremento en los TG en plasma176. Estos resultados concordarían con los observados en ratones ovariectomizados, donde la administración conjunta de estradiol y fenofibrato no tenía efectos aditivos, sino que más bien la hormona parecía suprimir la acción hipolipemiante del fibrato83. Por otro lado, para pacientes con diabetes que tienen hipertriacilglicerolemia, parece ser que el gemfibrocilo es el tratamiento más efectivo; sin embargo, no todos los pacientes toleran bien el tratamiento con este fármaco174. De cualquier manera, uno de los casos más evidentes es el descrito en individuos afectados por hipertriacilglicerolemia masiva que responden poco o nada al tratamiento con fibratos, con PUFA o con la asociación de fibratos y PUFA178. Realmente, en estos casos de hipertriacilglicerolemia extrema puede requerirse el tratamiento combinado de gemfibrocilo y niacina para reducir los TG a valores aceptables174. Curiosamente, el tratamiento con coenzima Q10 mejora la eficacia del fenofibrato en los pacientes con hipertriacilglicerolemia masiva que no responden a fenofibrato, posiblemente debido a un efecto directo en la mitocondria, con el incremento de la oxidación de los ácidos grasos, lo que consecuentemente provoca una reducción de los valores circulantes de FFA179.
Una observación que se repite en los pacientes tratados con fibratos es la variación considerable que se produce en los cambios lipídicos inducidos por el tratamiento. Se ha visto que los valores del PPARα en el hígado humano pueden varíar más de 2 veces entre diferentes individuos, por lo que es posible que esa respuesta interindividual diferente al tratamiento con fibratos se deba a los diferentes valores de expresión del PPARα43. No obstante, los diversos polimorfismos descritos en el gen del PPARα pueden contribuir también a la diferente respuesta al tratamiento con fibratos50. De hecho, se ha encontrado un efecto dependiente del genotipo en la respuesta al tratamiento con gemfibrocilo180, así como con fenofibrato181, en los valores de cHDL, que se ha relacionado con el polimorfismo Leu162Val del PPARα. La sustitución de leucina por valina (Leu162Val) que se ha descrito en el gen del PPARα da lugar a un receptor funcional, pero afecta a su activación transcripcional por ligando, según la concentración de éste182. El polimorfismo Leu162Val afecta al DBD del PPARα, de manera que el receptor mutante no puede unirse a los PPRE de forma tan eficiente como el receptor normal (wild-type). Sin embargo, para la forma mutante del receptor, la pobre activación que se observa con dosis bajas de ligando se normaliza e incluso aumenta a dosis elevadas, en comparación con la forma wild-type183. Por ende, se ha indicado que el efecto del polimorfismo Leu162Val del PPARα en la concentración de TG plasmáticos depende de la ingesta de PUFA en la dieta182. Por otro lado, se dividió a los pacientes de un estudio, sometidos al tratamiento durante 3 años con fenofibrato, en muy sensibles o poco sensibles al tratamiento, tomando como indicador el grado de TG plasmáticos. En los pacientes muy sensibles al fenofibrato, había una prevalencia elevada de ho-mocigotos GG en el intrón 7 del gen del PPARα, si se comparaba con los individuos de respuesta bajabb0920, bb0925. Se desconocen los mecanismos por los cuales la mutación en el intrón 7 puede influir en la disminución de los TG en respuesta al fenofibrato. Hay numerosos ejemplos de regiones reguladoras situadas en la zona de los intrones que afectan a la expresión del ARNm y los valores de proteína. Sin embargo, parece poco probable que la variante en el intrón 7 del gen del PPARα resulte por sí misma funcional, ya que no está cercana a la secuencia del sitio de ramificación para el ayuste (splicing)184. Entonces queda por dilucidar si el polimorfismo G/C del intrón 7 está directamente implicado en el control genético del valor de PPARα, o si solamente es un marcador de desequilibrio de ligamiento con una variante reguladora del gen PPARα. Por otro lado, se ha descrito que hay una relación entre las variaciones genéticas frecuentes de genes que codifican por proteínas relacionadas con el metabolismo de las lipoproteínas ricas en TG y la efectividad del fenofibrato en disminuir los valores de TG plasmáticos en individuos hipertriacilglicerolémicos. De este modo, los portadores de la mutación no sinónima (missense) Pro207Leu de la LPL, así como los de la Thr94Ala de la L-FABP presentan una hipertriacilglicerolemia residual después del tratamiento con fibratosbb0905, bb0930. En cuanto al polimorfismo del gen de la apo E, el alelo apo E2 está asociado con una respuesta mejor al fenofibrato en todos los parámetros lipídicos cuando se compara con las variantes E3 y E4. Es más, la presencia simultánea de apo E2 y del polimorfismo Leu162Val del PPARα tiende a mejorar la respuesta al fenofibrato en los individuos heterocigotos Pro207Leu para LPL181.
En algunos estudios clínicos con variables cardiovasculares como criterios de evaluación, los fibratos presentaron bien efectos beneficiosos, pero restringidos a ciertos tipos de pacientes, bien resultados negativos (recientemente revisado por Sanguino et al164). De acuerdo con esto, en el estudio Helsinki Heart Study (HHS), la reducción del riesgo de enfermedad cardiovascular después del tratamiento con gemfibrocilo era más pronunciada en los pacientes obesos con síndrome metabólico o diabetes y dislipemia aterogénicabb0815, bb0935. En el ensayo clínico Bezafibrate Infarction Prevention (BIP), la reducción de casos coronarios después del tratamiento con bezafibrato se observó tan sólo en los pacientes con valores elevados de TG en plasma. En el estudio clínico Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial (VAHIT), el efecto beneficioso significativo del gemfibrocilo se observó en pacientes diabéticos o en no-diabéticos con valores altos de insulina48. Es más, en el estudio clínico Fenofibrate Intervention and Event Lowering in Diabetes (FIELD), aunque el fenofibrato lograba disminuir tanto los valores de TG como los de cLDL, e incrementaba ligeramente el cHDL, no era capaz de reducir el riesgo de presentar episodios coronarios. Realmente, debido a la heterogeneidad de los individuos de estos estudios, las conclusiones realmente útiles se obtuvieron tras analizar los resultados de un subgrupo especial dentro la población total de pacientes164. Es más, en algunas ocasiones la interpretación de los estudios resultaba complicada, debido a que se usaban varios fármacos a la vez; por ejemplo, en el estudio FIELD, se utilizó de manera progresiva durante el estudio diferentes estatinas en los 2 grupos de estudio (placebo y fenofibrato)bb0815, bb0940. Por otro lado, se debería tener también en cuenta la influencia de otros factores, como la edad o la presencia de polimorfismos, en la efectividad de los fibratos. Así, por ejemplo, el análisis a posteriori de los datos del estudio FIELD, así como en el estudio Low Extremity Arterial Disease Event Lowering in Diabetes (LEADER), mostró una marcada disminución en los episodios cardiovasculares en la población de pacientes menores de 65 años, lo que indicaba que los pacientes mayores de 65 años eran los que no respondían a la administración del fibrato, y se perfilaba como una de las causas posibles de los resultados negativos obtenidos en estos estudios clínicos con fibratos164. Este resultado estaría además en consonancia con lo observado en animales viejos165. Es posible que se llegara a conclusiones similares si en estos estudios se examinase la posible asociación entre las variantes del gen PPARα y la respuesta al tratamiento con fibratos. Por ejemplo, en el estudio St. Mary's, Ealing, Northwick Park Diabetes Cardiovascular Disease Prevention (SENDCAP), se observó una reducción mayor de colesterol total en respuesta al bezafibrato en los portadores del alelo PPARα Val162 que en los que presentaban la variante Leu162184. Relacionado con esto, en el estudio VA-HIT se ha observado que la asociación entre el polimorfismo PPARα Leu162Val y el riesgo de presentar un episodio cardiovascular, así como la respuesta al tratamiento con gemfibrocilo, son más evidentes en los pacientes con resistencia a la insulina o diabetes mellitus189. Además, es necesario tener en cuenta que las primeras generaciones de fibratos (gemfibrocilo y bezafibrato) que se usaron en los estudios clínicos más antiguos son menos específicas para el PPARα que los fibratos de generaciones posteriores, como el fenofibrato, y que, por lo tanto, aquéllos actuarían, al menos en parte, también a través de PPARγ y/o PPARβ184. Finalmente, parece ser que compuestos del tipo β-caroteno son capaces de inhibir la activación del PPARα por sus agonistas58. Es más, como se ha comentado anteriormente, la coenzima Q10 también afecta a la efectividad del fenofibrato a la hora de corregir la dislipemia179. Por lo tanto, los resultados de los ensayos clínicos con fibratos podrían verse afetados por la suplementación que se haya usado: β-caroteno, coenzima Q10, etc.
Metabolismo de la glucosa y sensibilidad insulínicaEn diversos estudios, en los que se usaron ratones modificados genéticamente, se ha mostrado también una acción ineficaz de los activadores del PPAR, como por ejemplo los realizados con ratones obesos diabéticos (db/db). En estos animales, el tratamiento con rosiglitazona o Wy-14,643 normalizaba la expresión de genes relacionados con la homeostasia glucídica, lipídica y con la activación local de glucocorticoides. Sin embargo, un gran número de alteraciones de genes debidas a la diabetes permanecieron sin ser afectadas o incluso resultaron empeoradas por el tratamiento con los agonistas del PPAR. Así, la expresión alterada de muchos genes implicados en lipogenia, y en el funcionamiento peroxisomal y mitocondrial, no mejoró con el tratamiento, lo cual indica que las alteraciones moleculares asociadas a la diabetes las corrigen los agonistas del PPAR tan sólo de forma parcial190. Por otra parte, en ratas Zucker, la administración de fenofibrato durante 4 semanas no modificó los valores de insulina en suero, mientras que rosiglitazona sí conseguía disminuir estos valores de forma acusada167.
Es bien sabida la complejidad del papel del PPARα en la regulación de la sensibilidad a la insulina. En general, unos valores circulantes elevados de ácidos grasos y TG normalmente acompañan y/o inducen la resistencia a la insulina191. Algunos estudios han indicado un efecto beneficioso de la activación del PPARα en la sensibilidad insulínica, tanto en ratas normales como en modelos de resistencia insulínica de origen nutricional (dieta rica en grasa), genético (ratas Zucker obesas), o bien de tipo lipoatrófico (ratones A-ZIP/F-1)bb0715, bb0960, bb0965. En claro contraste con todo esto, la eliminación genética del PPARα no tuvo efectos en la sensibilidad insulínica194. De este modo, ratones carentes de PPARα alimentados con una dieta estándar no muestran signo alguno de resistencia insulínica, a pesar de presentar un aumento marcado en los FFA y TG circulantes191. Es más, se ha observado que los ratones carentes de PPARα se encuentran protegidos de desarrollar resistencia insulínica tras recibir una dieta grasa o simplemente en el proceso de envejecimiento194. Por el contrario, otros autores que han empleado el método de pinzamiento euglucémico hiper-insulinémico han observado que la deficiencia de PPARα no protege contra la resistencia insulínica inducida por una dieta grasa191. Es más, en otro estudio se observó que la activación farmacológica del PPARα exclusivamente en el cerebro conseguía reducir la utilización de glucosa por otros tejidos, mientras que en ratones carentes de PPARα la elevada utilización periférica de glucosa no disminuía por la reexpresión hepática del receptor195. Ello indicaría que el PPARα participa realmente en la regulación de la sensibilidad insulínica, aunque lo realiza a través de mecanismos que no requieren la expresión hepática de PPARα195.
Los estudios en humanos de los efectos de los fibratos en la sensibilidad insulínica han mostrado que inducen mejoras o que no tienen efecto, aunque probablemente se deba a la heterogeneidad de los individuos estudiados196. El factor predominante en pacientes obesos con síndrome metabólico es una elevación permanente de los FFA en plasma. De acuerdo con la hipótesis original de Randle et al197, el aumento en la disponibilidad de ácidos grasos por el músculo inhibe competitivamente la oxidación de glucosa, y provoca una disminución en la captación de glucosa y, así, resistencia insulínica. La activación del PPARα favorece la oxidación de ácidos grasos, y es una de las razones por las que los fibratos ejercen una acción hipolipemiante y la causa de que los ligandos del PPARα puedan, en algunas situaciones, mejorar la sensibilidad insulínica al reducir la acumulación de lípidos en los tejidos193. Sin embargo, sólo unos pocos estudios clínicos han descrito una mejoría en la homeostasia glucídica tras el tratamiento con fibratosbb0990, bb0995. Si bien los efectos beneficiosos de los fibratos en el control glucídico puede que no sean atribuibles totalmente al aumento en la oxidación de los ácidos grasos relacionado con la activación del PPARα, ya que alguno de esos agentes es también capaz de activar ligeramente al PPARγ200. Por el contrario, el estudio clínico FIELD no reveló efecto alguno del fenofibrato en los parámetros glucídicos de pacientes diabéticos201. Es más, en un artículo reciente se concluye que la administración de fenofibrato durante 3 meses a pacientes obesos con diabetes mellitus tipo 2 no afectó a su sensibilidad insulínica202. Más aún, Subramanian et al203 han descrito que la activación del PPARα en individuos sanos no obesos no es capaz de revertir la resistencia insulínica inducida por la administración de glucocorticoides.
Inflamación y efectos vascularesAunque diversos estudios han mostrado las propiedades antiaterogénicas de los ligandos del PPARα in vitro, los modelos en ratones muestran resultados contradictorios48. Aquí, de nuevo, la bibliografía muestra incongruencias entre los efectos de los agonistas del PPARα y el fenotipo del ratón carente de PPARα. Sorprendentemente, los ratones carentes de PPARα y de apo E desarrollan menos lesiones ateroscleróticas cuando se alimentan con una dieta grasa que los ratones carentes tan sólo de apo E, lo que indica un papel aterogénico del PPARαbb0300, bb1020. Estos ratones carentes de PPARα/apo E muestran una concentración mayor de lipoproteínas aterogénicas y una presión sanguínea menor204. Esta menor presión sanguínea en los animales carentes de PPARα puede contribuir a reducir el número de lesiones ateroscleróticas observadas en estos animales, a pesar de la presencia en ellos de valores elevados de lipoproteínas aterogénicas205.
Los efectos de los fibratos en la enfermedad vascular no están todavía dilucidados196. Algunos estudios han mostrado que el papel antiinflamatorio del PPARα en la pared vascular parece ser dependiente de la gravedad de la inflamación o de la lesión vascular. Así, en ausencia de inflamación o en el estadio inicial de la lesión, los efectos del PPARα son principalmente proaterogénicosbb1030, bb1035, mientras que el desarrollo de lesiones graves, acompañadas de inflamación, se ve reducido claramente por la activación del PPARα. En ese sentido, aunque la expresión de la proteína quimioatrayente de monocitos (MCP-1, monocyte chemoattractant protein), una quimiocina que promueve la quimiotaxis de los monocitos, es claramente inhibida por los ligandos del PPARγ, el efecto de los activadores del PPARα no está claro. Es más, se ha observado que en células endoteliales aórticas humanas, los ligandos naturales y sintéticos del PPARα estimulan la síntesis de MCP-1206. Por el contrario, Pasceri et al208 han encontrado que los fibratos reducen la expresión de MCP-1 inducida por la PCR en células endoteliales de vena umbilical humana. Además, como ya se ha comentado, el PPARα participa en la inhibición de la síntesis de moléculas proinflamatorias, como interleucinas y prostaglandinas, mediante la disminución de la actividad del NF-κB209. Por el contrario, el tratamiento con agonistas del PPARα incrementa la concentración del factor de necrosis tumoral alfa (TNF-α), un agente proinflamatorio, inducido en plasma por LPS. Curiosamente, ese efecto es menor en ratones carentes de PPARα210, lo que indica un papel proinflamatorio del PPARα. Por el contrario, Poynter y Daynes209 han observado que los esplenocitos de ratones carentes de PPARα producen valores mayores de IL-6, lo que apoya una acción antiinflamatoria dependiente del PPARα.
Otro potencial efecto inesperado del tratamiento con fibratos a través del PPARα es un aumento en la homocisteína plasmática (un factor de riesgo emergente) en pacientes con dislipemia, que podría reducir además la producción hepática de apo A-I196. Aunque el aumento en la homocisteína puede contrarrestar el esperado efecto cardioprotector del fibrato, los datos del estudio Diabetes Atherosclerosis Intervention Study (DAIS) apuntan a que el aumento en los valores de homocisteína observados tras el uso de fibratos no atenúan su efecto beneficioso en la aterosclerosis coronaria163.
Soluciones farmacológicas y nutricionalesEl síndrome metabólico, la diabetes mellitus tipo 2 y la dislipemia asociada a este tipo de enfermedades son alteraciones cuya incidencia está aumentando en todo el mundo. Por ende, muchos de estos pacientes no están recibiendo el tratamiento apropiadobb0885, bb1055. Por lo general, se ha aceptado el uso de fibratos, agonistas del PPARα, como el tratamiento más prometedor212. Sin embargo, como ya se ha comentado, hay diversos estudios, tanto en animales como en humanos, que muestran que la activación del PPARα puede tener efectos potencialmente adversos o que los fibratos pueden fallar, lo que hace muy necesaria la búsqueda de tratamientos complementarios o alternativosbb0010, bb0290. Además, los fibratos son agonistas débiles del PPARα, por lo que son necesarias dosis elevadas de fármaco para un tratamiento efectivo. Todo ello hace suponer que agonistas del PPARα más selectivos, con más potencia y especificidad, presentarán una efectividad mayor y menos efectos adversos. Estos tratamientos alternativos se están desarrollando actualmente, y aún no se ha establecido su utilidadbb0010, bb0090, bb1065.
Por otra parte, cualquiera que sea el tratamiento mediado por PPARα que se aplique, debería basarse en una comprensión mejor de la biología de los PPAR, de la naturaleza de los agonistas endógenos de PPAR, y de cómo se generan estas moléculas. Es más, cuando las rutas que sirven para producir los agonistas endógenos de PPAR fallan, puede que estén generando ligandos inadecuados para PPAR, que contribuirían a la aparición de situaciones patológicas58. Si éste fuera realmente el caso, una estrategia alternativa podría basarse en promover la producción de agonistas endógenos “apropiados” o en el diseño de agentes que pudieran competir y desplazar a los ligandos “inadecuados” en su unión al PPAR. De hecho, hay una hipótesis que propone que los agonistas del PPARγ, las TZD, actuarían mediante un mecanismo de ese tipobb0010, bb1070.
Agonistas duales del PPARα/γ y PPARα/δ y panagonistasLos agonistas duales del PPAR se desarrollan con la idea de combinar la capacidad de los agonistas del PPARα (fibratos) para reducir los valores de TG plasmáticos con la mejora de la sensibilidad insulínica que promueven los agonistas del PPARγ (TZD)215. De hecho, en modelos animales se ha demostrado que la combinación de fibratos y agentes sensibilizadores a la insulina produce, a través de mecanismos complementarios, una disminución en la resistencia insulínica, la glucemia y los valores de TG plasmáticos216, así como en la inflamación vascular217. Por lo tanto, es de suponer que un agente que simultáneamente active PPARα y PPARγ será útil para el tratamiento de esas disfunciones del metabolismo215.
Los glitazars son agonistas duales de PPARα y PPARγ que se están investigando en el tratamiento de la diabetes mellitus y el síndrome metabólico. Entre otros, están naveglitazar, muraglitazar, ragaglitazar, tesaglitazar y chiglitazarbb1075, bb1090. Todos ellos reducen los valores de TG y aumentan los de las HDL, con lo que se mejora la sensibilidad insulínica. Además, ragaglitazar, tesaglitazar y muraglitazar mejoran el metabolismo de ácidos grasos y lipoproteínas al disminuir los valores plasmáticos de apo B y apo C-III e inducir la actividad hepática de LPL y CPT-1213. Además, el tratamiento con ragaglitazar reduce de forma significativa la presión sanguínea en ratas espontáneamente hipertensas y mejora la función endotelial en ratas Zucker obesas cuando se compara con el tratamiento con pioglitazona219. Estudios farmacológicos in vivo en ratones controles (no obesos) y en diabéticos db/db demuestran que muraglitazar modula la expresión de genes diana de PPAR, y muestran una acción potente y eficaz como antidiabético y como agente hipolipemiante220. Debido a los excelentes hallazgos encontrados en modelos animales, se iniciaron diversos estudios clínicos en humanos. En un estudio clínico en fase III con pacientes con diabetes mellitus tipo 2, se comparó el tratamiento con muraglitazar frente a pioglitazona, ambos fármacos en combinación con metformina221. En comparación con el efecto de pioglitazona y metformina, el tratamiento con muraglitazar y metformina provocó una mejoría más evidente en el control glucémico, una reducción mayor en los TG plasmáticos y un incremento más acusado en las HDL plasmáticas. Sin embargo, el tratamiento con muraglitazar provocó un número mayor de muertes, infartos no fatales de miocardio o derrames cerebrales que en el caso de los pacientes tratados con pioglitazona o con placebo222. Aún más, en estudios clínicos se ha observado que el tratamiento con tesaglitazar eleva los valores en sangre de creatinina, lo que indica una potencial toxicidad renal. Por lo tanto, el uso de estos 2 prometedores compuestos se suspendió en mayo de 2006, y actualmente se están estudiando otras moléculas nuevas222.
Por otro lado, DRF 2519, un análogo de las TZD, también presenta activación dual de PPARα y γ. En el modelo de ratón resistente a la insulina ob/ob, el DRF 2519 consiguió una mayor mejora de la resistencia insulínica y dislipemia que la rosiglitazona. En ratas Zucker obesas, el DRF 2519 mostró una reducción mayor de la insulina plasmática, y de los valores de TG y ácidos grasos, así como un aumento de la relajación de la musculatura lisa aórtica, en comparación con rosiglitazona223. En ratas Sprague–Dawley alimentadas con una dieta grasa, el DRF 2519 mejoró el perfil lipídico plasmático de manera más clara que fenofibrato o rosiglitazona. Estos resultados indican que DRF 2519 podría ser un candidato interesante en el tratamiento de trastornos metabólicos y las complicaciones asociadas223. Otro nuevo agonista dual del PPARα/PPARγ es LSN862, que mejora los valores de glucosa y de lípidos en modelos de roedores con diabetes mellitus tipo 2 y dislipemia. En ratas diabéticas Zucker obesas, el LSN862 mostró una eficacia mayor en reducir los valores de glucosa y TG que la rosiglitazona. Además, en ratones db/db, el LSN862 demostró una mejor eficacia antidiabética si se comparaba con rosiglitazona. En ratones transgénicos portadores de apo A-I humana, el LSN862 y el fenofibrato reducían los valores de VLDL, mientras que rosiglitazona los aumentaba224. Además, un nuevo agonista dual PPARα/γ, el (S)-3-(4-[2 carbazol-9-il-etoxi] fenil-2-etoxi-ácido propiónico), ha demostrado que produce una mejora en la sensibilidad insulínica225. Otros nuevos agonistas duales del PPARα/γ, que no son de tipo TZD y que se han desarrollado recientemente, son C333H y T33, los cuales presentan actividad sensibilizadora a la insulina e hipolipemiantebb1130, bb1135. Por otro lado, el MK-767 (KRP-297), a diferencia de otras TZD, activa no sólo al PPARγ, sino también al PPARα228.
Actualmente, también se están desarrollando agonistas duales para PPARα y PPARδ. Es predecible que estos agonistas duales sean capaces de disminuir la hiperlipemia, resistencia insulínica y los factores de riesgo cardiovascular. Recientemente, se ha demostrado que el T659, un agonista dual del PPARα/δ, producía un aumento en los valores de HDL en primates229. El interés de este hallazgo es que hay muy pocos agentes terapéuticos capaces de aumentar las concentraciones de cHDL en pacientes dislipémicos, ya que los fibratos tan sólo causan un incremento ligero en las concentraciones de cHDL, y las TZD, al igual que las estatinas, tienen poco o ningún efecto en el cHDL.
El bezafibrato es el más conocido de los panagonistas (α, β y γ) de PPAR, con más de un cuarto de siglo de empleo terapéutico seguro. El bezafibrato mejora tanto la sensibilidad insulínica como el perfil lipídico plasmático en pacientes con síndrome metabólico230. Por lo tanto, los estudios clínicos basados en el bezafibrato apoyan el uso de ligandos panPPAR como agentes terapéuticos contra el síndrome metabólico. No obstante, ya que el bezafibrato es un ligando débil de PPAR, se deberían desarrollar nuevos compuestos pan-PPAR más poderosos para el tratamiento de enfermedades en las que coexistan alteraciones del metabolismo lipídico y glucídico231. En este sentido, netoglitazona y PLX 204 son nuevos ligandos panagonistas que se encuentran actualmente en investigación. En un futuro próximo, esos ligandos podrían ser potentes agentes terapéuticos para el tratamiento de diabetes asociada con complicaciones cardiovascularesbb1075, bb1160. De hecho, los panagonistas del PPAR han mostrado su actividad antidiabética sin la ganancia de peso que sí se observa con el uso de agonistas del PPARγ215. De este modo, netoglitazona producía una disminución de los valores plasmáticos de ácidos grasos, glucosa y TG, así como un aumento en la sensibilidad insulínica en ratas obesas y diabéticas233. No obstante, debido a los potenciales múltiples sitios de acción de los pan-PPAR, que afectarían a la transcripción de una gran cantidad de genes diferentes, resulta imprescindible una información más amplia con respecto a su toxicidad en humanos215.
Moduladores selectivos de PPAR (SPPARM) y agonistas parcialesEl concepto de moduladores selectivos de PPAR (SPPARM) fue sugerido por analogía con el término de moduladores selectivos del receptor de estrógenos (SERM), a partir de que cada ligando diferente puede tener propiedades agonistas o antagonistas distintas, dependiendo del contexto celular y del gen diana específico de que se trate. Los ligandos del PPAR con diferentes estructuras químicas (SPPARM) se unen al LBD del receptor, e inducen cambios conformacionales distintos que conllevan diferentes afinidades de unión para los diversos cofactores y que, por tanto, activan al PPAR de distintas maneras, provocando efectos diferenciales en la expresión génica y en las respuestas biológicas. No obstante, debemos tener en mente que, a diferencia de otros receptores clásicos de hormonas esteroideas, los receptores PPAR tienen un gran bolsillo de unión al ligando, que no se rellena completamente con el ligando234. Según esto, la hipótesis de trabajo es que la mejor estrategia terapéutica sería conseguir una acción beneficiosa mediada por el PPAR en un tipo celular, sin inducir un efecto adverso mediado también por el PPAR, en otro46. La gran mayoría de los SPPARM desarrollados hasta la fecha actúan en el PPARγ y han demostrado que modifican selectivamente la expresión génica, así como reducen la resistencia insulínica sin causar ganancia de peso215. Así, en ratones diabéticos, metaglidasen disminuye las concentraciones de insulina y glucosa, aumenta la concentración de adiponectina y mejora el perfil lipídico plasmático sin modificar el peso215. El FK614 es un SPPARM con propiedades diferenciales en la regulación del funcionamiento de la célula grasa. Este compuesto se comporta como agonista parcial cuando induce al PPARγ a interaccionar con los coactivadores CBP y SRC-1, pero actúa como agonista total con PBP y PRIP, que se requieren para la adipogenia mediada por PPARγ. Por tanto, el FK614 induce la expresión del ARNm para aP2 y la acumulación de TG en adipocitos 3T3-L1 que se estén diferenciando, pero no así en adipocitos maduros, mientras que las TZD producen esos mismos efectos en los 2 estadios de diferenciación de los adipocitos. Es por ello que, dado que FK614 se comporta como un SPPARM con una selectividad dependiente del estadio de diferenciación celular, podría disminuir la resistencia insulínica, sin estimular la acumulación de grasa en los adipocitos235. Por otro lado, el gemfibrocilo, un agonista clásico del PPARα, recientemente se ha considerado como un SPPARM43. Ello estaría en consonancia con el hecho de que el gemfibrocilo es estructuralmente único en comparación con el resto de fibratos236. De ese modo, mientras el fenofibrato se comporta como un agonista total, el gemfibrocilo actuaría como un modulador selectivo del PPARα, induciendo una selección diferente de los coactivadores por el complejo PPARα:RXR-ADN, según el PPRE de que se trate43. Cada posible interacción diferente entre el PPARα y sus cofactores generaría señales distintas que producirían una actividad reguladora “característica” de la expresión de genes66. Estas observaciones podrían explicar las diferencias encontradas entre fenofibrato y gemfibrocilo en el efecto que ejercen en la expresión y los valores plasmáticos de apo A-I43.
Es de reseñar que Feige et al237 han establecido claramente la diferencia entre moduladores selectivos del PPAR y los agonistas parciales de PPAR, 2 términos que con demasiada frecuencia se intercambianbb0215, bb1075. Los SPPARM inducen cambios conformacionales distintos a los de los agonistas totales y provocan una inducción selectiva de la selección de correguladores, de la activación de genes diana y de los efectos fisiológicos. Realmente, los SPPARM difieren de los agonistas parciales en que promueven la regulación selectiva de genes a través de la diferente modulación de la transcripción de los genes diana, específica para cada gen. De este modo, algunos genes son inducidos a valores similares a los observados con agonistas totales, mientras que otros muestran una activación más limitada. Por contra, los agonistas parciales provocan una activación global de todos los genes diana, más reducida que la producida por los agonistas totales237.
Otros tratamientos farmacológicosLa represión del PPAR es y ha sido también un campo de estudio en el desarrollo de tratamientos hipolipemiantes, antidiabetogénicos y antiinflamatorios. Ello proviene de la hipótesis de que muchos ligandos sintéticos de PPARγ no son puros activadores de PPARγ, sino que, más bien, actúan como agonistas parciales. De hecho, se ha propuesto que estas moléculas competirían con ligandos endógenos, los cuales sí que se comportarían como agonistas puros, por unirse al PPAR. Así pues, las TZD son capaces de incrementar la sensibilización a la insulina mediante el desplazamiento de ligandos endógenos del PPARγ, que estarían afectando negativamente a genes glucorreguladores clave y a la acción de la insulina214. Por lo tanto, revertiendo esta represión con un agonista parcial del tipo TZD, se estaría aumentando la sensibilidad a la insulina. De hecho, ese efecto se ha comprobado en los ratones heterocigotos PPARγ+/−, con la reducción de la presencia del propio receptor PPARγ214. No obstante, la acción de los TZD más aceptada es que inducen la activación del PPARγ, por lo que los TZD se consideran, por lo general, agonistas de PPARγ. Por contra, el compuesto SR-202 sí que se ha identificado como antagonista del PPARγ. Curiosamente, tanto SR-202 como TZD protegen de la resistencia a la insulina inducida por una dieta grasa en modelos animales, lo que indica que los agonistas y antagonistas de PPARγ tienen efectos similares en roedores. Sin embargo, al contrario que las TZD, SR-202 reduce la adipogenia y previene la aparición de obesidad238.
Sin embargo, para el caso del PPARα no se cumple este comportamiento, ya que se ha observado que el antagonismo en el PPARα produce efectos similares a los descritos cuando se inhibe al PPARα, es decir, fallos en la regulación del metabolismo lipídico y lipoproteico239.
Por otra parte, los ligandos para RXR (rexinoides) se han considerado también como posibles agentes terapéuticos para usarlos en enfermedades metabólicas. Un heterodímero compuesto por RXR y PPAR puede ser activado por un ligando del RXR, independientemente de si está o no presente el ligando para el PPAR. Es más, los homodímeros de RXR pueden unirse también a PPRE funcionales e inducir la transactivación, tanto en presencia como en ausencia de PPAR240. De hecho, los ligandos sintéticos de tipo rexinoide, como la targretina (LGD1069), pueden activar genes que contengan un PPRE en su promotor de una manera independiente de la formación del complejo PPAR-RXR241. Así, en ratón, los rexinoides han mostrado efectos antidiabéticos242, pero, por contra, también incrementaron los valores de TG circulantes243 e interfirieron en la ruta de señalización del TR, provocando un intenso hipotiroidismo244.
Una combinación de agentes hipolipemiantes, que se ha usado en pacientes dislipémicos con respuesta baja a la monoterapia, ha sido la de fibratos con el ácido nicotínico (niacina), el cual, a pesar de su modesto efecto incrementando la glucemia, muestra efectos beneficiosos en los tradicionales parámetros lipídicos y fracciones lipoproteicas en sangre, y produce una reducción importante en los episodios cardiovasculares. Posiblemente, los efectos beneficiosos observados en los valores de FFA se deban a la inhibición que ejerce la niacina en la lipólisis del tejido adiposobb1225, bb1230. Así por ejemplo, la niacina se ha combinado con el clofibrato en una molécula, el etofibrato, que ha demostrado tener efectos hipolipemiantes tanto en animales247, como en personas248.
NutracéuticaLos cambios inducidos por la dieta en los valores circulantes de ácidos grasos insaturados pueden ser suficientes para modificar de una forma apreciable la actividad del PPAR18. Además, el ácido eicosapentanoico y el docosahexanoico, ligandos naturales del PPARα se pueden encontrar frecuentemente en los suplementos de aceite de pescado o de ω-3, que se pueden adquirir sin receta215. Es interesante señalar que los PUFA, especialmente los de la clase ω-3, pueden afectar a todos los receptores nucleares que regulan los valores de TG, esto es, LXR, HNF-4α y los 3 subtipos de PPAR249, lo que explicaría su eficiente efecto hipotriacilglicerolemiante. Los ácidos grasos ω-3 son activadores de PPARα más potentes que los de la serie ω-6, pero ninguno de los 2 tipos de ácidos grasos son realmente activadores potentes de PPARα. Por el contrario, sus derivados de tipo eicosanoide, o bien los ácidos grasos oxidados, tienen una gran afinidad por el PPARα y son activadores potentes tanto de la isoforma alfa como de la gammabb1250, bb1255.
El ácido linoleico conjugado (CLA) se encuentra de forma natural en tejidos animales y en la dieta, así como también en carnes de rumiantes, de aves de corral, en huevos y en productos lácteos. Aunque los mecanismos de acción de CLA no están completamente claros, se ha propuesto que la activación del PPARα y/o PPARγ puede contribuir, al menos en parte, en los efectos beneficiosos de CLA en aterogenia, diabetes y la función inmunitaria, que se han observado en diversos estudios animalesbb1260, bb1265.
Las amidas de los ácidos grasos con etanolamina (FAE) se producen de forma natural en las células de mamíferos a través de la acción concertada de 2 enzimas. Los FAE son una familia de lípidos de origen natural que son mediadores activos, están presentes en tejidos y en sangre, y participan en diversas funciones biológicas, como son la regulación del apetito254 y la mejora de la función car-díaca en condiciones de cardiomiopatía255. Generalmente, actúan a través de la activación de proteínas G acopladas a receptores de cannabinoides. Sin embargo, se ha demostrado que la FAE que porta ácido oleico, llamada oleiletanolamida (OEA), estimula la actividad lipolítica del tejido adiposo y la oxidación hepática de ácidos grasos mediante un mecanismo independiente de los receptores de cannabinoides, y que implica la activación del PPARα256.
La disminución en la expresión del PPARα podría contribuir al estado prooxidante que se observa en animales envejecidos, y que posiblemente es causado por una deficiente modulación del estado rédox celular. Así, se ha observado que los ratones viejos expresan valores reducidos de los ARNm para PPARα y sus genes diana, como ACO peroxisómica. De manera recíproca, ese estado prooxidante podría ser causado por la expresión reducida del PPARα encontrada en ratas viejas165. De acuerdo con ello, en ratones viejos se ha constatado que la suplementación con vitamina E provoca un aumento en los ARN transcritos de PPARα y ACO, alcanzando los valores observados en animales jóvenes209. Ello indicaría que, si se equilibra de forma adecuada el estado rédox celular, se puede actuar de forma positiva en la regulación transcripcional del PPARα. Además, dado que algunos de los efectos adversos de la acción del PPARα parecen deberse a los compuestos generados en el metabolismo de oxidación de los ácidos grasos257, la coadministración de un tratamiento antioxidante, como la vitamina E, junto con ligandos del PPAR, podría aliviar o prevenir los efectos secundarios de los agonistas del PPAR46.
Hay algunos estudios acerca de la actividad biológica de las isohumulonas encontradas en la cerveza, y en algunos de ellos se ha descrito que estos compuestos pueden activar a PPARα y PPARγ258. Es más, se ha propuesto que las isohumulonas mejoran la resistencia insulínica en ratones diabéticos mediante el aumento de la expresión del PPARγ258, mientras que el aumento en los valores plasmáticos de cHDL y la reducción de la concentración de colesterol y de TG en hígado de ratón podría deberse al aumento que inducen en la expresión del PPARα259. Un mecanismo similar podría explicar el efecto de las isohumulonas en la prevención de la obesidad inducida por la dieta en ratones260.
En diversos estudios epidemiológicos y nutricionales realizados en humanos y en animales, se ha indicado que el consumo de proteína de soja reduce los valores plasmáticos de colesterol total, cLDL y TG, así como el contenido hepático de colesterol y Tg261. La proteína de soja es única entre todas las proteínas de origen vegetal, porque se asocia con isoflavonas262. Así, Mezei et al263, usando ratones carentes de PPARα alimentados con dietas que contenían diferentes cantidades de isoflavonas de soja, determinaron que la mejoría significativa observada en los valores de TG plasmáticos se debía a la ingesta de las isoflavonas de la soja y que se producía a través de mecanismos tanto dependientes como independientes del PPARα. Curiosamente, la estructura química de las isoflavonas es similar a la de los fibratos264. De hecho, estudios in vitro han mostrado que las isoflavonas de la soja, particularmente genisteína y daidzeína, son capaces de activar tanto PPARα como PPARγ263. En cultivos celulares, se ha demostrado que tanto los extractos de soja que contienen isoflavonas, como las isoflavonas de soja por separado, son capaces de incrementar la expresión de genes diana de PPAR262. Por otro lado, Mezei et al265 mostraron que el consumo de proteína de soja rica en isoflavonas mejora la tolerancia a la glucosa, la resistencia a la insulina y las concentraciones de colesterol y TG hepáticos en ratas Zucker obesas.
Como se ha comentado anteriormente, el heterodímero compuesto por RXR y PPAR puede ser activado por el ligando del RXR, ácido 9-cis retinoico, independientemente de la presencia o ausencia de un ligando para PPAR. El ácido retinoico puede formarse mediante la ruptura simétrica del β-caroteno. No obstante, el β-caroteno se puede también escindir de manera asimétrica y originar una serie de moléculas conocidas como apocarotenales. Recientemente se ha hipotetizado con la idea de que los apocarotenales, como el ácido 9-cis retinoico, po-drían actuar como moduladores de la transcripción. Así, diversos estudios han revelado que el β-apo-14' carotenal (apo14) es capaz de inhibir la activación de PPARα y RXR producida por sus respectivos agonistas. Entonces, un posible papel del apo14 podría ser modular la activación del PPARα58.
Finalmente, se ha observado que compuestos polifenólicos, como el resveratrol y sus análogos, que se encuentran en uvas, vino y cacahuetes, pueden actuar como hipolipemiantes e hipoglucemiantes actuando a través del PPARα266. Rimando et al266 han investigado si este compuesto y sus 3 análogos (pterostilbeno, piceatannol y resveratrol trimetil éter) podrían activar al PPARα in vivo e in vitro. Así, se ha constatado que el pterostilbeno presentaba la máxima inducción del PPARα de los 4 compuestos. De acuerdo con esto, en hámsters hipercolesterolémicos, el pterostilbeno disminuyó los valores plasmáticos de cLDL y glucosa, y aumentó los valores plasmáticos de cHDL, cuando se comparó con el grupo control.
ConclusionesSon necesarios, de manera urgente, fármacos efectivos y bien tolerados para controlar la obesidad, las enfermedades cardiovasculares, la diabetes mellitus tipo 2 y, en definitiva, el síndrome metabólico. Para conseguirlo, se necesitan más estudios y ensayos clínicos que ayuden a completar nuestro conocimiento sobre cómo el PPARα modula de forma intracelular y extracelular las rutas metabólicas glucídicas y lipídicas, y cómo regula las actividades inflamatorias vascular y hepática. Es más, hay numerosos efectos pleiotrópicos de los fibratos que participan en la acción beneficiosa de los agonistas del PPARα en la dislipemia, más allá del impacto positivo que estos agentes puedan tener en el metabolismo lipídico y lipoproteico. Estos y otros efectos de los agonistas del PPARα que están por des-velar podrán ayudarnos a conseguir mejorar su efectividad y a desarrollar nuevas moléculas que puedan usarse como tratamiento en el síndrome metabólico, la diabetes mellitus tipo 2 y la dislipemia.
Agradecimientos
Queremos expresar nuestro agradecimiento a la Sra. Milagros Morante por su excelente ayuda técnica.
* Este estudio ha sido financiado por el Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica (I+D+i), del Instituto de Salud Carlos III-Subdirección General de Evaluación y Fomento de la Investigación (PI060352) así como por la Fundación Universitaria San Pablo-CEU (USP-PC 16/07).
Recibido 11 Febrero 2008
Aceptado 11 Marzo 2008
Dr. C. Bocos. Facultad de Farmacia. Universidad San Pablo-CEU. Urbanización Montepríncipe. 28668 Boadilla del Monte. Madrid. España. carbocos@ceu.es