





La LDL electronegativa (LDL[−]) es una fracción minoritaria de la LDL total que se encuentra en circulación y presenta diferentes propiedades inflamatorias, siendo una de las más relevantes la inducción de citoquinas en células endoteliales y mononucleares. Sin embargo, no se conoce el mecanismo por el cual la LDL(−) ejerce su acción a nivel celular. El objetivo de este estudio fue evaluar los posibles receptores implicados en la unión de la LDL(−) en monocitos, como consecuencia de la cual se induciría la liberación de citoquinas.
MétodosSe valoró la liberación de MCP1, IL6 e IL10 como citoquinas representativas inducidas por la LDL(−) en monocitos. Se evaluó la producción de estas citoquinas en la condición basal, LDL(−) sola, y las condiciones en que se inhibía los diferentes receptores selectivamente mediante anticuerpos.
ResultadosSe obtuvo que la LDL(−) compite con el LPS en la producción de citoquinas y parece hacerlo por las vías de los receptores toll-like (TLR) TLR2 y TLR4 asociadas a CD14. Los resultados indican que los proteoglicanos (PG) también podrían tener algún papel, pero sólo en la liberación de MCP1 y el receptor CD36 en la de IL6.
Marcando la LDL(−) fluorescentemente se realizaron estudios de unión a los monocitos, obteniéndose que la LDL(−) se une a las células aproximadamente igual que la LDL(+) en cuanto a cantidad de partículas. En esta unión total ni los PG ni el CD36 serían vías de entrada mayoritarias, ya que su bloqueo no afectó a dicha unión. En cambio con anticuerpos antiCD14 y antiTLRs se inhibió significativamente la unión de LDL(−) a monocitos.
ConclusionesEn la inducción de citoquinas por LDL(−) en monocitos están implicados algunos de los receptores estudiados, entre ellos parece importante la vía TLR-CD14, aunque sería necesario profundizar en el papel de este receptor y en su implicación en la acción de la LDL(−).
Elecronegative LDL (LDL(−)) is a small fraction of the total circulating LDL and has different inflammatory properties, one of the most important being the induction of cytokines in endotelial cells and monocytes. However, the mechanism by which LDL (−) exercises its action at cellular level is not known.
The objective of this study was to evaluate the receptors involved in LDL (−) binding in monocytes, and how the liberation of cytokines is induce.
MethodsThe liberation of MCP1, IL6 and IL10 were evaluated as representative cytokines induced by LDL(−) in monocytes. The production of these cytokines was assessed under baseline conditions, LDL(−) only, and under conditions in which it is inhibited by different receptors using antibodies.
ResultsIt was shown that LDL (−) competes with LPS in the production of cytokines and appears to do this via the toll like receptors (TLR), TLR2 and TLR4 associated with CD14. The results indicate that the proteoglycans (PG) could also play a role, but only in the liberation of MCP1and the CD36 receptor in IL6 release.
Monocyte binding studies using fluorescent labeled LDL(−) were performed, which showed that LDL(−) bound to the cells almost as much as LDL(+) as regards the number of particles. In this total binding, neither the PG nor the CD36 could be major entry points, since their block did not affect this binding. On the other hand, the binding of LDL(−) to monocytes was significantly inhibited with anti-CD14 and anti-TLRs antibodies.
ConclusionIn conclusion, some of the receptors studied are involved in cytokine induction by LDL(−) in monocytes, among them the TLR-CD14 pathway appears to be important, although the role of this receptor and its involvement in the action of LDL(−) needs to be studied in more detail.
La LDL electronegativa (LDL[−]) es una fracción minoritaria (1–10%) de la LDL total, la cual, debido a su mayor carga negativa, es aislable por cromatografía de intercambio aniónico. Existen discrepancias entre los autores respecto a algunas de las propiedades de esta partícula, sin embargo hay acuerdo en que la LDL(−) presenta propiedades aterogénicas como son una densidad y tamaño alterados1, una menor afinidad por el receptor de LDL2, una mayor agregabilidad3, mayor unión a proteoglicanos (PG)4 y actividad fosfolipolítica aumentada5. Entre las acciones más relevantes inducidas por la LDL(−) en células endoteliales se encuentra la inducción de toxicidad y apoptosis6,7, así como la liberación de citoquinas inflamatorias8,9. Además, en el caso de monocitos y linfocitos se ha descrito que la LDL(−) induce diversos genes de citoquinas inflamatorias pero también antiinflamatorias como la IL10, así como Fas cuya expresión en membrana se encuentra aumentada10,11.
A pesar del interés que suscita la existencia de una LDL modificada en circulación con propiedades aterogénicas, apenas se conocen los mecanismos que dan lugar a la formación de dichas partículas ni cuales son los receptores celulares o las vías de transmisión que participan en los efectos de la LDL(−) en las células. Respecto a la implicación de factores de transcripción en el efecto mediado por LDL(−), nuestro grupo ha descrito que en células endoteliales se activan NF-kB y AP-112 que coincide con resultados de otros autores13. Sin embargo, en células mononucleares la LDL(−) no parece inducir AP-1 y tampoco los PPAR, aunque sí parece estar implicado el NF-kB10.
En el aspecto de los receptores celulares, se conoce que la LDL(−), en comparación con la LDL(+), presenta menor afinidad por el receptor de LDL (rLDL) de fibroblastos y no es captada a través del receptor scavenger (SR) del tipo AII de macrófagos2. Otros datos de que se dispone están centrados en el modelo de células endoteliales en cultivo, en el caso de células progenitoras la LDL(−) promovió senescencia a través del receptor celular LOX-114. Sin embargo, los monocitos no expresan LOX y no se conocen los receptores implicados en la unión y subsiguiente efecto de la LDL en ese tipo celular. Por tanto, el objetivo del presente estudio fue evaluar algunos receptores candidatos y su relación con la acción inflamatoria de la LDL(−) en monocitos. Concretamente, se analizó el papel de los PG, los receptores toll-like 2 y 4 (TLR2, TLR4), el CD14 y el CD36.
En cuanto a los PG, sabemos que la LDL(−) presenta una mayor afinidad que la forma nativa especialmente por el condroitín sulfato (CS)4, que a su vez es el más abundante en la superficie de monocitos. Se ha descrito que lipoproteínas unidas a PG pueden ser internalizadas por vías como la fagocitosis15, por lo que la LDL(−) podría ser captada a través de esta vía, preferentemente a la LDL(+), e inducir su efecto inflamatorio. El CD36 es otro posible candidato, el cual es un SR que reconoce lipoproteínas nativas y modificadas y se encuentra expresado en monocitos y macrófagos16.
Por otra parte, los TLRs son receptores con un papel importante en la respuesta immunológica innata a patógenos, ya que reconocen componentes de la pared bacteriana, como el lipopolisacárido (LPS) y, en algunos casos, como TLR2 y TLR4, también reconocen lipoproteínas17. Se ha descrito que los TLRs participan en el proceso aterogénico, hallándose una elevada expresión de TLR2 y TLR4 en el desarrollo de la lesión, tanto en placa aterosclerótica como en células circulantes18. Por otra parte, en estudios de interacción entre TLRs y LDL modificada o fosfolipídos oxidados, se ha hallado que dicha unión puede comportar la inducción de citoquinas de forma dependiente o independiente de CD1419. CD14 es el principal receptor de LPS que tras la unión del ligando se asocia principalmente a TLR4, aunque también puede hacerlo a TLR2, y esto provoca la transducción de señales intracelulares que activan la expresión de genes inflamatorios20. Se ha descrito que CD14 también puede unir lipoproteínas, como la LDL mínimamente oxidada (LDLmm)21.
En este trabajo se ha evaluado la implicación de los mencionados receptores como posibles responsables de la unión de la LDL(−) en monocitos. Este conocimiento sería de gran importancia, ya que nos aportaría a su vez datos sobre las posibles vías activadas a nivel intracelular que podrían ser las promotoras de la liberación de citoquinas en este tipo celular.
MétodosAislamiento de LDL total y subfraccionamiento en LDL(+) y LDL(−)La LDL total (rango de densidad 1.019–1.050g/ml) se aisló a partir de mezclas de plasma, dado el elevado número de determinaciones a realizar y la cantidad de muestra necesaria para ello. Se utilizó plasma-EDTA a partir de voluntarios normolipémicos (NL) con concentraciones de colesterol total<6,5mmol/l, colesterol LDL<4,0mmol/l y triglicérido<2mmol/l. Todos los sujetos eran normoglicémicos, no fumadores y con una edad comprendida entre 30–60 años. Se hicieron mezclas de plasma de 10–15 individuos con el fin de obtener un volumen de plasma de 100–150ml. La separación de la LDL total se llevó a cabo mediante ultracentrifugación secuencial y a densidad de 1.050g/ml. A partir de la LDL total se aislaron las fracciones LDL(+) (o LDL nativa) y LDL(−) mediante cromatografía de intercambio aniónico en una columna preparativa Hiload-Q-Sepharose 26/10 en un sistema AKTA-FPLC (Pharmacia), siguiendo metodología descrita3. Posteriormente se concentraron las fracciones por ultracentrifugación y se conservaron a 4°C hasta el momento de realizar las determinaciones, las cuales se desarrollaron en el periodo de 1 semana.
Aislamiento de monocitos a partir de plasmaEl aislamiento de monocitos a partir de plasma de voluntarios sanos se realizó en función de la densidad según el protocolo utilizado previamente en estudios ya publicados11. Los leucocitos se sembraron en placas de 12 pocillos a la densidad de 1,5 millones de células por pocillo. Al día siguiente del aislamiento celular se procedió a descartar los linfocitos, que al no presentar propiedades adhesivas y quedar en suspensión en el medio de cultivo, se pueden separar fácilmente de los monocitos que quedan adheridos. Los monocitos se cultivaron en las condiciones óptimas de crecimiento, con medio RPMI-10% suero bovino fetal (SBF) a 37°C y 5% de CO2.
Estudios de captación de LDL marcada fluorescentementePara los estudios de internalización celular, la LDL(+) y LDL(−) se marcaron con el compuesto fluorescente DiI22 (LDL(+)-DiI, LDL(−)-DiI) y en todos los experimentos se determinó la actividad específica de marcaje. Los monocitos se incubaron con la LDL marcada durante 3h a 37°C y la captación celular se determinó mediante técnicas fluorimétricas descritas por nuestro grupo2. Se realizaron estudios de desplazamiento de la unión de LDL(+) o LDL(−) marcada (50mg apoB/l) con concentraciones crecientes de LDL(+) y LDL(−) sin marcar (50–200mg apoB/l), para comprobar cual es captada con mayor avidez por las células. También se usó como ligando competidor del desplazamiento LPS a concentraciones crecientes (10–1000μg/l).
Por otra parte, se hicieron experimentos con concentraciones fijas de LDL(+) y LDL(–) marcadas (50mg apoB/L) en ausencia o presencia de compuestos que bloquean la unión a los receptores a estudiar. En algunos casos se trataron los monocitos con forbol miristato acetato (PMA) (50μg/l) 24h antes de la incubación con las LDLs para diferenciarlos a macrófagos.
Para evaluar la unión a PG se usó la digestión enzimática de glicosaminoglicanos (GAG) y competición con GAG libres23. Concretamente se pretrataron las células durantes 2h con condroitinasa ABC (500U/L y 2000U/L) antes de añadir las LDLs, o bien se adicionó condroitin-3-sulfato (CS) (0,3 y 0,6g/l) a los monocitos simultáneamente a las LDLs.
Se utilizaron anticuerpos antiCD36, a concentraciones de 5 y 10mg/l, antiCD14, antiTLR2 y antiTLR4, a 2 y 5mg/l, para bloquear dichos receptores. Los anticuerpos fueron añadidos a los monocitos 1 hora antes de la adición de las LDLs.
Estudios de bloqueo de receptores y liberación de citoquinasEl objetivo de estos experimentos fue bloquear la entrada de diferentes receptores celulares candidatos a unir LDL(−) y evaluar si se inhibía la liberación de citoquinas inducida por la LDL(−). Los monocitos fueron pretratados con los mismos compuestos para bloquear receptores descritos en el apartado anterior y en las mismas condiciones. Para llevar a cabo los experimentos las LDL fueron dializadas en medio deficiente (1% SBF), filtradas en esterilidad y adicionadas al cultivo a una concentración de 150mg apoB/l. La incubación de los monocitos con LDL(+) y LDL(−) y con los diferentes inhibidores de receptores y vías celulares, fue de 4 y 20h. Después de la incubación se recogió el sobrenadante celular y conservó a −80°C hasta la determinación de las citoquinas liberadas (IL6, IL10 y MCP1) mediante ELISA según lo descrito11.
En la liberación de citoquinas de cada condición se restó la producción basal, obtenida por la adición a los monocitos de los compuestos o anticuerpos correspondientes. Se usó LPS (0,1mg/l) como control positivo de inducción de citoquinas y se ensayó la condición de coincubación de las células con LPS y LDL(+) o LDL(−).
ResultadosUnión total de LDL(+) y LDL(−) a monocitosSe marcó fluorescentemente con DiI LDL(+) y LDL(−), obteniendo que el marcaje de la LDL(−) siempre fue inferior al de la LDL(+) como indica su actividad específica (tabla 1), probablemente debido a causas estructurales y/o agregación de la partícula. Sin embargo, en los experimentos con células, al interpolar los valores de fluorescencia en la recta de actividad específica de cada subfracción, se obtiene que en monocitos se une la misma cantidad de LDL(−) que de LDL(+), tal y como se observa en la tabla 1. En la tabla se muestran los valores obtenidos con 50mg/l de LDL marcada e incubación con monocitos a 37°C. También se valoró la unión a 4°C y diferentes concentraciones de LDL, sin observarse diferencias en los resultados.
Actividad específica de LDL(+) y LDL(–) marcada con DiI y captación a monocitos de LDL-DiI a 50mg/l (n=6).
| Actividad específica (mg DiI/g DiI-LDL) | Captación celular (μg/l apoB) | |
| LDL(+) | 70.74±23.27 | 159.95±36.76 |
| LDL(−) | 25.91±25.67* | 157.88±57.17 |
Desplazando DiI-LDL(−) con LDL(+) se observó que la unión de ambos ligandos presenta igual afinidad (dato no mostrado), coincidiendo con los resultados de unión total. De todas formas, la ausencia de diferencias en el nivel de unión de LDL(+) y LDL(−) no es indicativo de que se unan al mismo tipo de receptores.
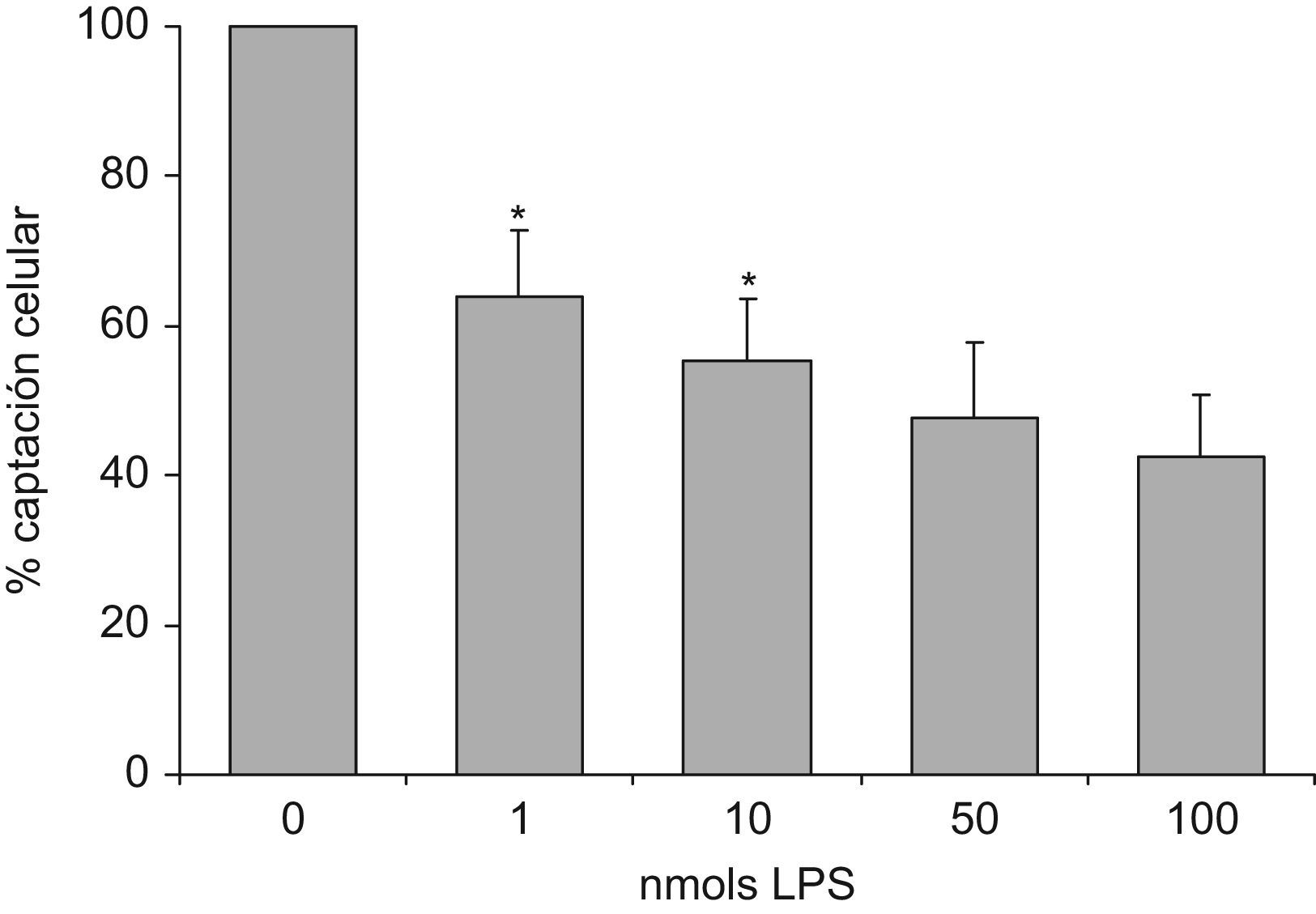
Si el ligando utilizado para desplazar era LPS se obtuvo que a la misma relación molar (100nmoles de LDL y LPS) el desplazamiento de la unión de LDL(−) fue sobre el 60% (fig. 1). De hecho, con bajas cantidades de LPS el desplazamiento de la unión de LDL(−) fue ya muy eficaz, ya que con una relación de moles de 100 veces menos de LPS respecto a LDL(−) ya desplazó casi el 40% de la captación celular de LDL(−). El aumento de la concentración de LPS en 100 veces sólo se tradujo en un incremento de la inhibición del 20%.
-DiI a monocitos en presencia de concentraciones crecientes de LPS. Se incubaron los monocitos con LDL(−)-DiI (50mg apoB/l=100nmols apoB/l) en ausencia de LPS y en presencia de concentraciones crecientes de LPS (0–100nmols LPS). Se valoró la captación a las 3h de incubación a 37°C por técnicas fluorimétricas. Los valores se expresaron considerando el 100% de unión la LDL(−)-DiI unida en ausencia de LPS. Los resultados son la media de 3 experimentos y se indicaron las significaciones estadísticas con * versus la unión a la anterior concentración menor de LPS.")
Desplazamiento de la unión de LDL(−)-DiI a monocitos en presencia de concentraciones crecientes de LPS. Se incubaron los monocitos con LDL(−)-DiI (50mg apoB/l=100nmols apoB/l) en ausencia de LPS y en presencia de concentraciones crecientes de LPS (0–100nmols LPS). Se valoró la captación a las 3h de incubación a 37°C por técnicas fluorimétricas. Los valores se expresaron considerando el 100% de unión la LDL(−)-DiI unida en ausencia de LPS. Los resultados son la media de 3 experimentos y se indicaron las significaciones estadísticas con * versus la unión a la anterior concentración menor de LPS.
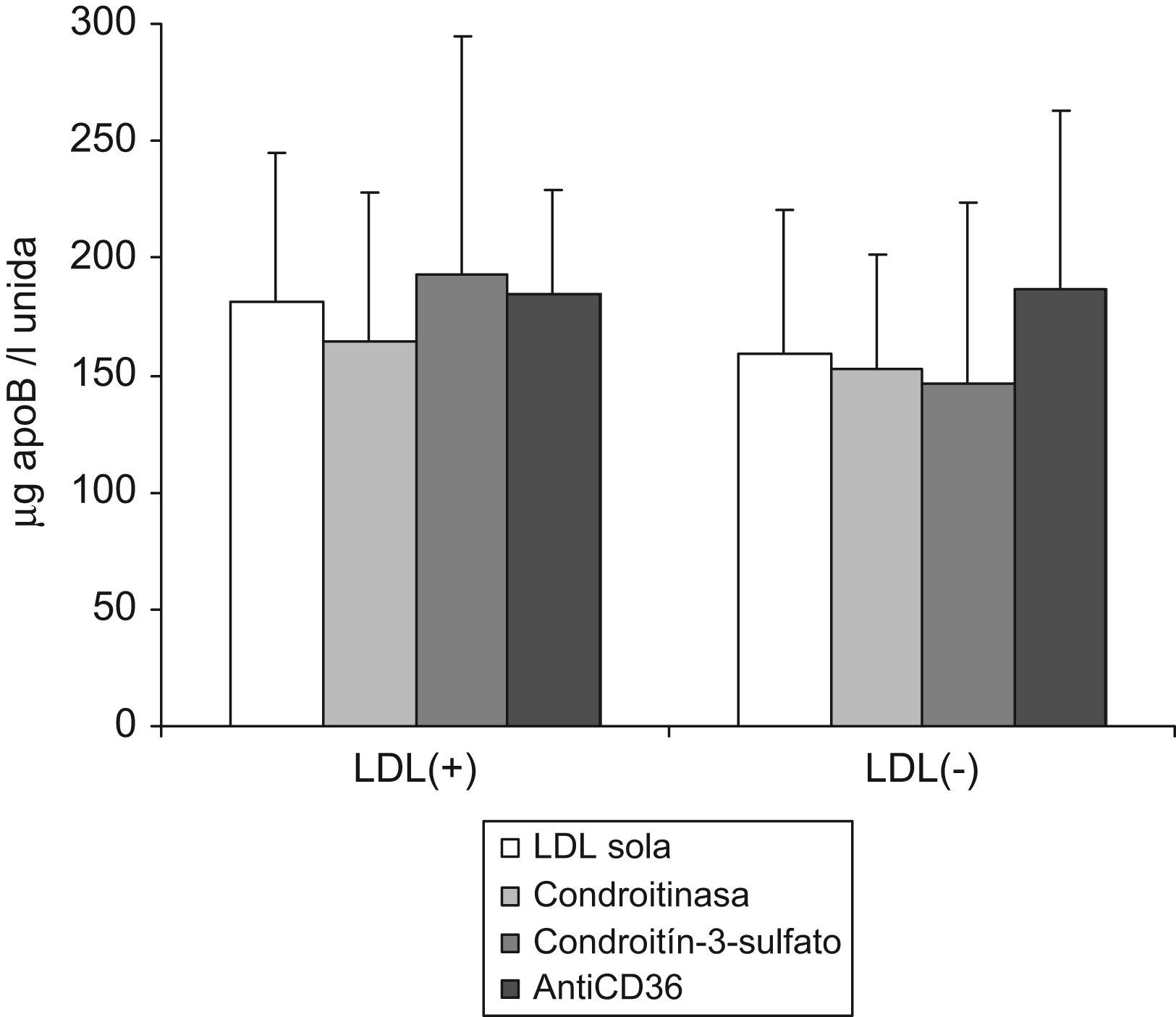
La LDL(+) y LDL(−) marcadas con DiI se incubaron con los monocitos que previamente habían estado sometidos a bloqueo de receptores y se comparó la unión celular respecto a la de los monocitos en situación basal. Como se observa en la figura 2, el bloqueo de la unión a PG, por tratamiento con condroitinasa ABC o condroitín sulfato, no afecta a la unión total de LDL(+) o LDL(−) a los monocitos, por tanto, los PG no sería una vía mayoritaria de entrada de las LDL a las células. El bloqueo de CD36 con un anticuerpo específico tampoco afectó a la unión total.
 o con condroitín sulfato (CS) (0,3g/l) o bien 1h con anticuerpo antiCD36 (5mg/l). Posteriormente se incubaron las células con LDL(−)-DiI o LDL(+)-DiI (50mg apoB/l) y se valoró su captación (μg apoB/l de LDL-DiI unida) en las diferentes condiciones: sin tratamiento previo (barras blancas), con tratamiento con condroitinasa (gris claro), con CS (gris oscuro) o con antiCD36 negro. Los resultados son la media de 4 experimentos.")
Captación de LDLs a los monocitos en presencia de bloqueantes de PG y CD36. Los monocitos se preincubaron 2h con condroitinasa ABC (500U/L) o con condroitín sulfato (CS) (0,3g/l) o bien 1h con anticuerpo antiCD36 (5mg/l). Posteriormente se incubaron las células con LDL(−)-DiI o LDL(+)-DiI (50mg apoB/l) y se valoró su captación (μg apoB/l de LDL-DiI unida) en las diferentes condiciones: sin tratamiento previo (barras blancas), con tratamiento con condroitinasa (gris claro), con CS (gris oscuro) o con antiCD36 negro. Los resultados son la media de 4 experimentos.
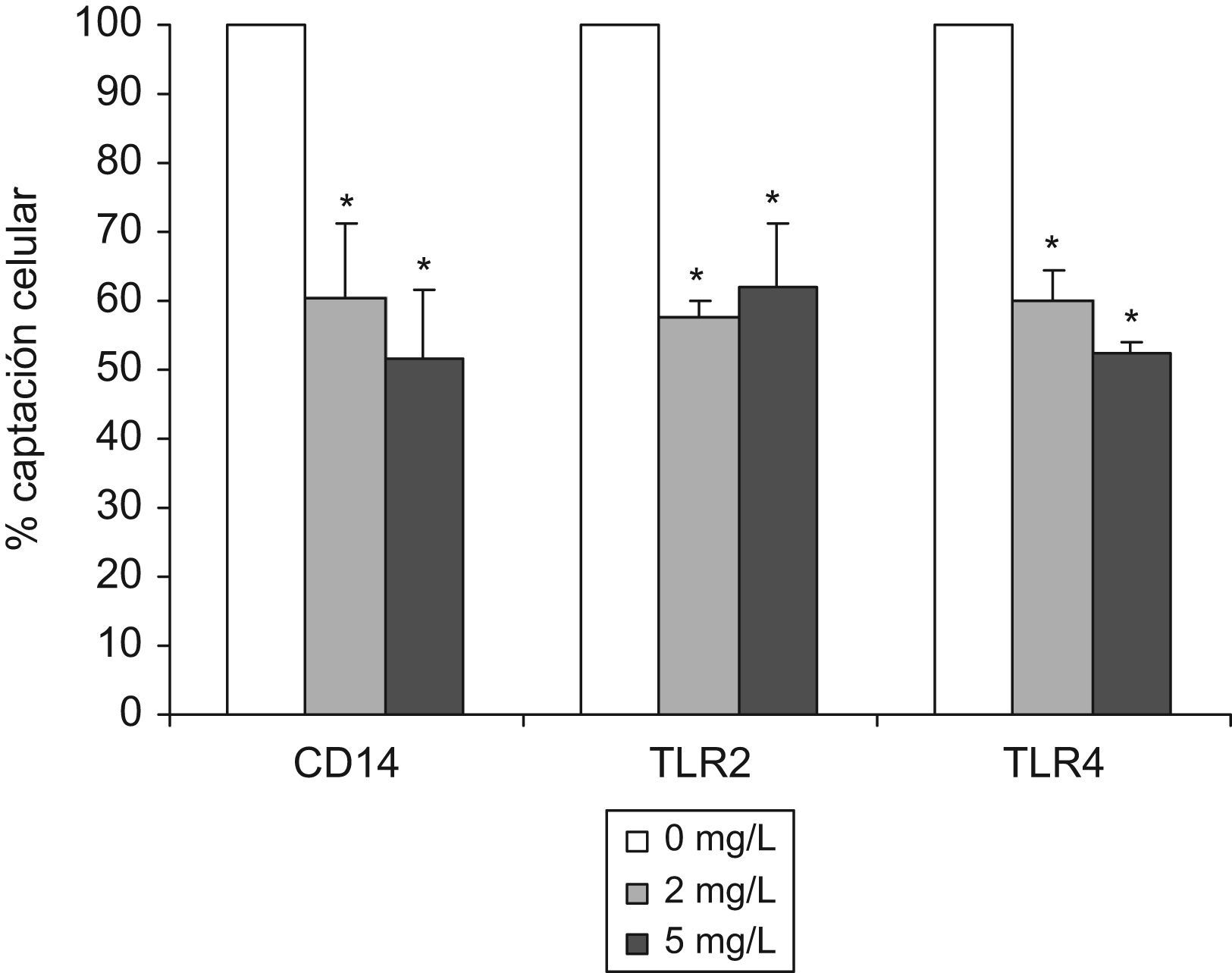
En cambio, el bloqueo de CD14 disminuyó la unión de LDL(−) a los monocitos (fig. 3), mientras que no afectó a la unión de la LDL(+) (dato no mostrado). Este mismo efecto se observó cuando se incubaron los monocitos con anticuerpos anti TLR2 y TLR4. El bloqueo de la unión de LDL(−) no se incrementó significativamente al aumentar la concentración de anticuerpo, así que con la concentración de 2mg/l ya se consigue prácticamente la máxima inhibición de la unión.
-DiI por monocitos en presencia de anticuerpos antiCD14, antiTLR2 y antiTLR4. Los monocitos se preincubaron 1h con anticuerpos antiCD14, antiTLR2 o antiTLR4, a 2 y 5mg/l, para bloquear dichos receptores. Posteriormente se incubaron las células con LDL(−)-DiI (50mg apoB/l) y se valoró la captación celular con los anticuerpos a 2mg/l (barras gris claro) y 5mg/l (gris oscuro) respecto a la condición en que no se hizo tratamiento (blanco). Los resultados son la media de 4 experimentos y se indican las significaciones estadísticas con * versus la unión con la LDL(−)-DiI en la condición en que no se ha hecho ningún tratamiento, con P=0,068.")
Captación de LDL(−)-DiI por monocitos en presencia de anticuerpos antiCD14, antiTLR2 y antiTLR4. Los monocitos se preincubaron 1h con anticuerpos antiCD14, antiTLR2 o antiTLR4, a 2 y 5mg/l, para bloquear dichos receptores. Posteriormente se incubaron las células con LDL(−)-DiI (50mg apoB/l) y se valoró la captación celular con los anticuerpos a 2mg/l (barras gris claro) y 5mg/l (gris oscuro) respecto a la condición en que no se hizo tratamiento (blanco). Los resultados son la media de 4 experimentos y se indican las significaciones estadísticas con * versus la unión con la LDL(−)-DiI en la condición en que no se ha hecho ningún tratamiento, con P=0,068.
No hubo diferencias en ninguno de estos resultados en la condición en que se habían diferenciado las células previamente con PMA.
Efecto de los PG y CD36 en la liberación de citoquinasEl hecho de que la unión total a la célula no se vea afectada por el bloqueo de los PG o de CD36 y que este comportamiento sea igual para LDL(+) y LDL(−), no implica que pueda haber una cierta unión a estos receptores que pueda desencadenar la liberación de citoquinas. Por tanto, se evaluó la producción de citoquinas cuando se bloqueaba los receptores a estudiar.
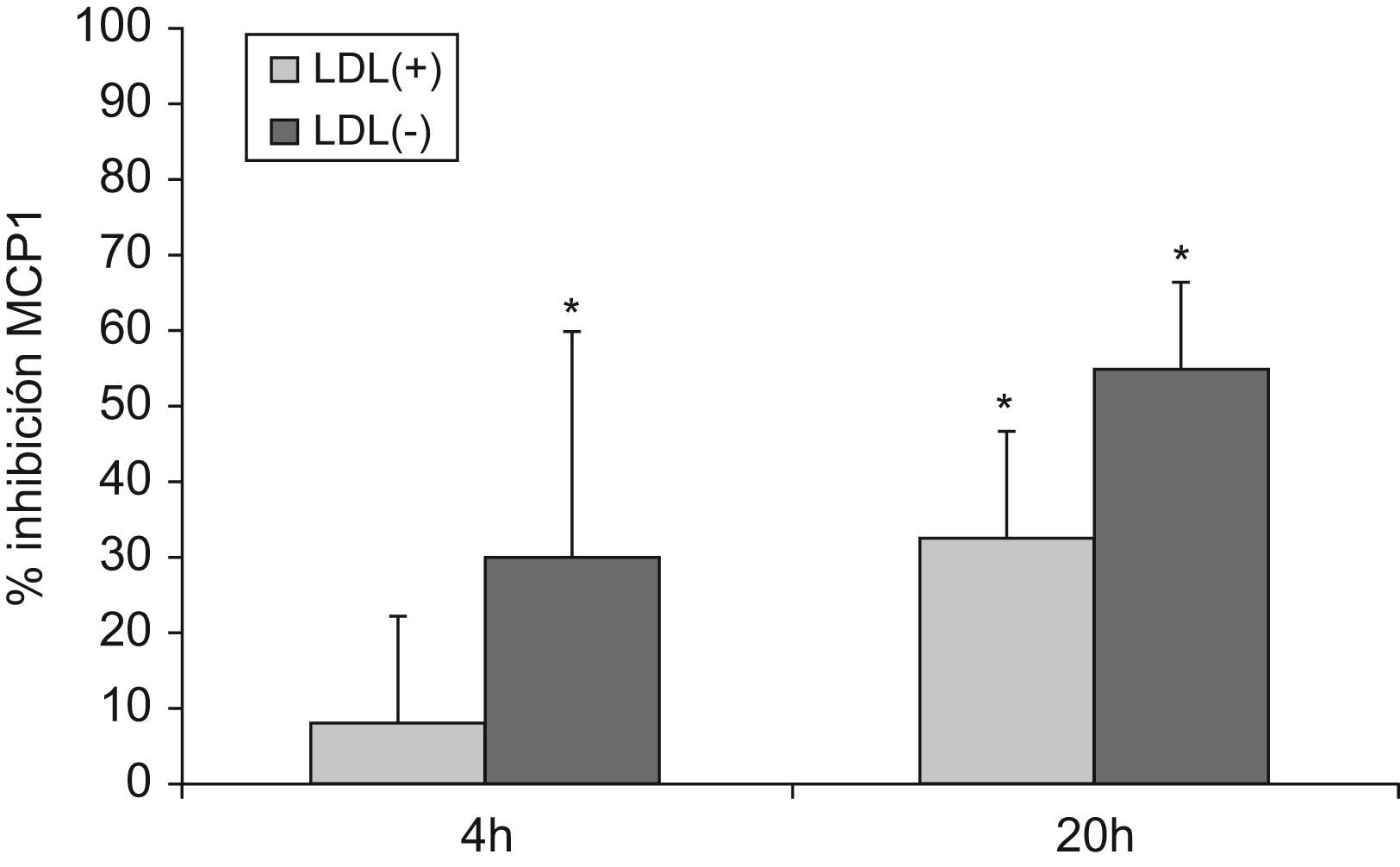
En la condición basal, sin lipoproteínas, se obtuvo un aumento en la liberación de citoquinas cuando se trataban los monocitos con condrotinasa, valor que fue restado en las condiciones en que se había añadido este enzima. Por otra parte, no se observaron diferencias en la liberación de IL6 ni IL10 por LDL(+) ni por LDL(−), a ninguna de las concentraciones utilizadas de condroitinasa ni a 4 o 20h de incubación con las LDLs (datos no mostrados). En cambio, sí hubo inhibición en la liberación de MCP1 como se observa en la figura 4 (500U condroitinasa/l), dicha inhibición fue mayor en el caso de la LDL(−) y más a las 20h de incubación con LDL(−) que a las 4h. Resultados similares se obtuvieron con el uso de una mayor concentración de condroitinasa (2000U/l).
 y LDL(−). Los monocitos se preincubaron 2h con condroitinasa ABC (500U/l). Posteriormente se incubaron las células con LDL(+) o LDL(−) (150mg apoB/l) (barras gris claro y/gris oscuro respectivamente) durante 4 o 20h y tras la incubación se recogió el sobrenadante celular para evaluar MCP1 mediante ELISA. Los resultados son el % de inhibición en la liberación de MCP1 respecto a la condición en que los monocitos fueron sólo tratados con LDL sin preincubación con condroitinasa. Se presenta la media de 4 experimentos y se indican las significaciones estadísticas con * versus la liberación inducida por la LDL en monocitos no preincubados con condroitinasa, con P=0,068.")
Inhibición de la liberación de MCP1 en monocitos tratados con condroitinasa previamente a la adición de LDL(+) y LDL(−). Los monocitos se preincubaron 2h con condroitinasa ABC (500U/l). Posteriormente se incubaron las células con LDL(+) o LDL(−) (150mg apoB/l) (barras gris claro y/gris oscuro respectivamente) durante 4 o 20h y tras la incubación se recogió el sobrenadante celular para evaluar MCP1 mediante ELISA. Los resultados son el % de inhibición en la liberación de MCP1 respecto a la condición en que los monocitos fueron sólo tratados con LDL sin preincubación con condroitinasa. Se presenta la media de 4 experimentos y se indican las significaciones estadísticas con * versus la liberación inducida por la LDL en monocitos no preincubados con condroitinasa, con P=0,068.
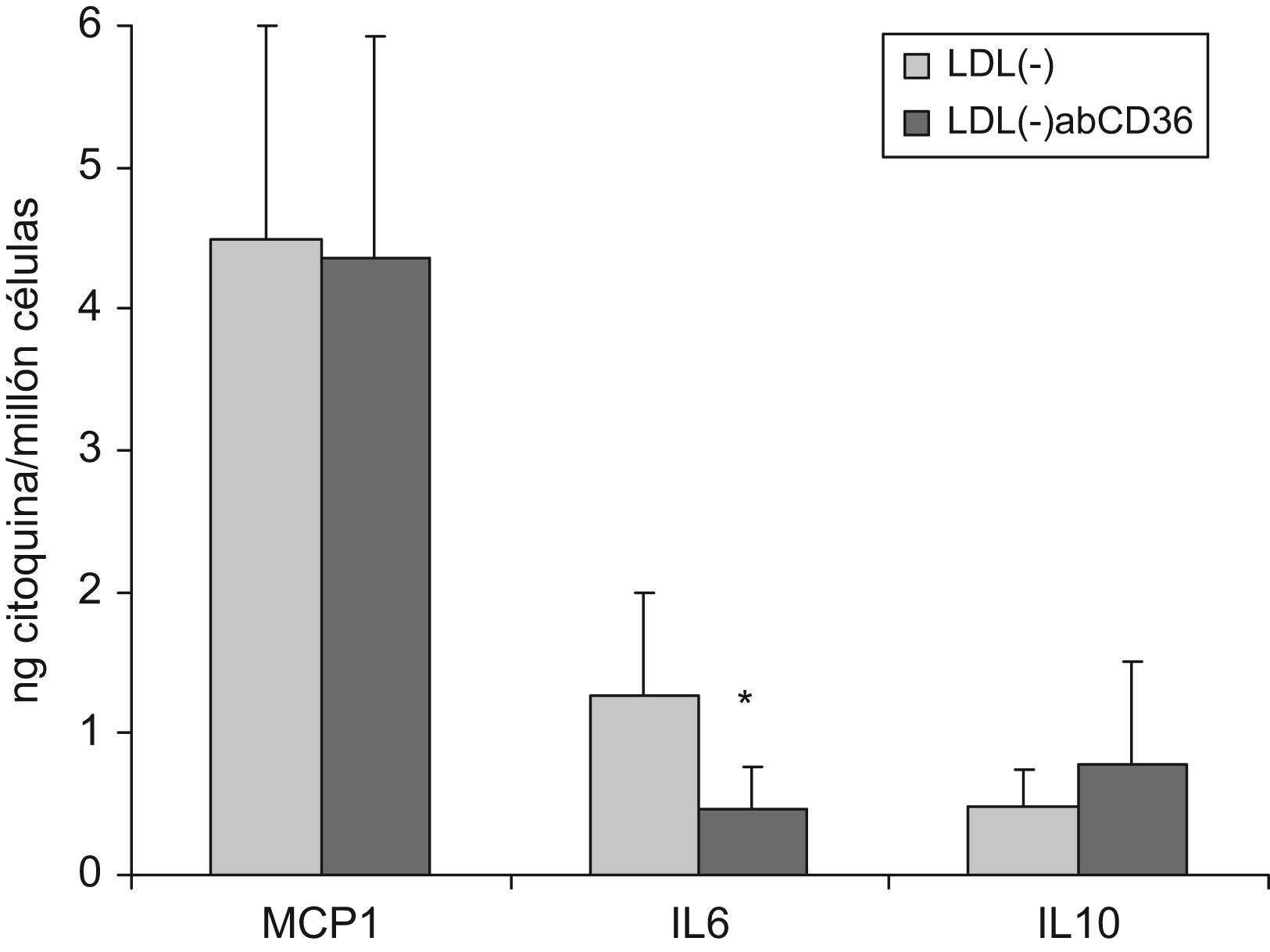
En el caso del bloqueo de CD36 no hubieron diferencias significativas en la liberación de MCP1 ni IL10, pero sí en el de IL6 por parte de LDL(−) (fig. 5) (5mg CD36/l), aunque no por LDL(+). Esto indicaría que el receptor CD36 podría estar implicado en la liberación de esa citoquina inducida por LDL(−). Estos resultados fueron similares a 4 y 20h de incubación y a una mayor concentración de CD36 (10mg/l). Tampoco se observaron cambios significativos cuando los monocitos habían sido diferenciados por estimulación con PMA.
. Los monocitos se preincubaron 1h con antiCD36 (5mg/l) previo a la adición de LDL(−) (150mg apoB/l) durante 20h y tras la incubación se recogió el sobrenadante celular para valorar las citoquinas mediante ELISA. Los resultados se expresan como ng de la citoquina correspondiente liberada por cada millón de células, en monocitos sin preincubar (barras gris claro) o con preincubación con anticuerpo antiCD36 (gris oscuro). Se presenta la media de 4 experimentos y se indican las significaciones estadísticas con * versus la liberación inducida por la LDL(−) en monocitos no preincubados con antiCD36, con P=0,068.")
Liberación de MCP1, IL6 e IL10 en monocitos tratados con antiCD36 previamente a la adición de LDL(−). Los monocitos se preincubaron 1h con antiCD36 (5mg/l) previo a la adición de LDL(−) (150mg apoB/l) durante 20h y tras la incubación se recogió el sobrenadante celular para valorar las citoquinas mediante ELISA. Los resultados se expresan como ng de la citoquina correspondiente liberada por cada millón de células, en monocitos sin preincubar (barras gris claro) o con preincubación con anticuerpo antiCD36 (gris oscuro). Se presenta la media de 4 experimentos y se indican las significaciones estadísticas con * versus la liberación inducida por la LDL(−) en monocitos no preincubados con antiCD36, con P=0,068.
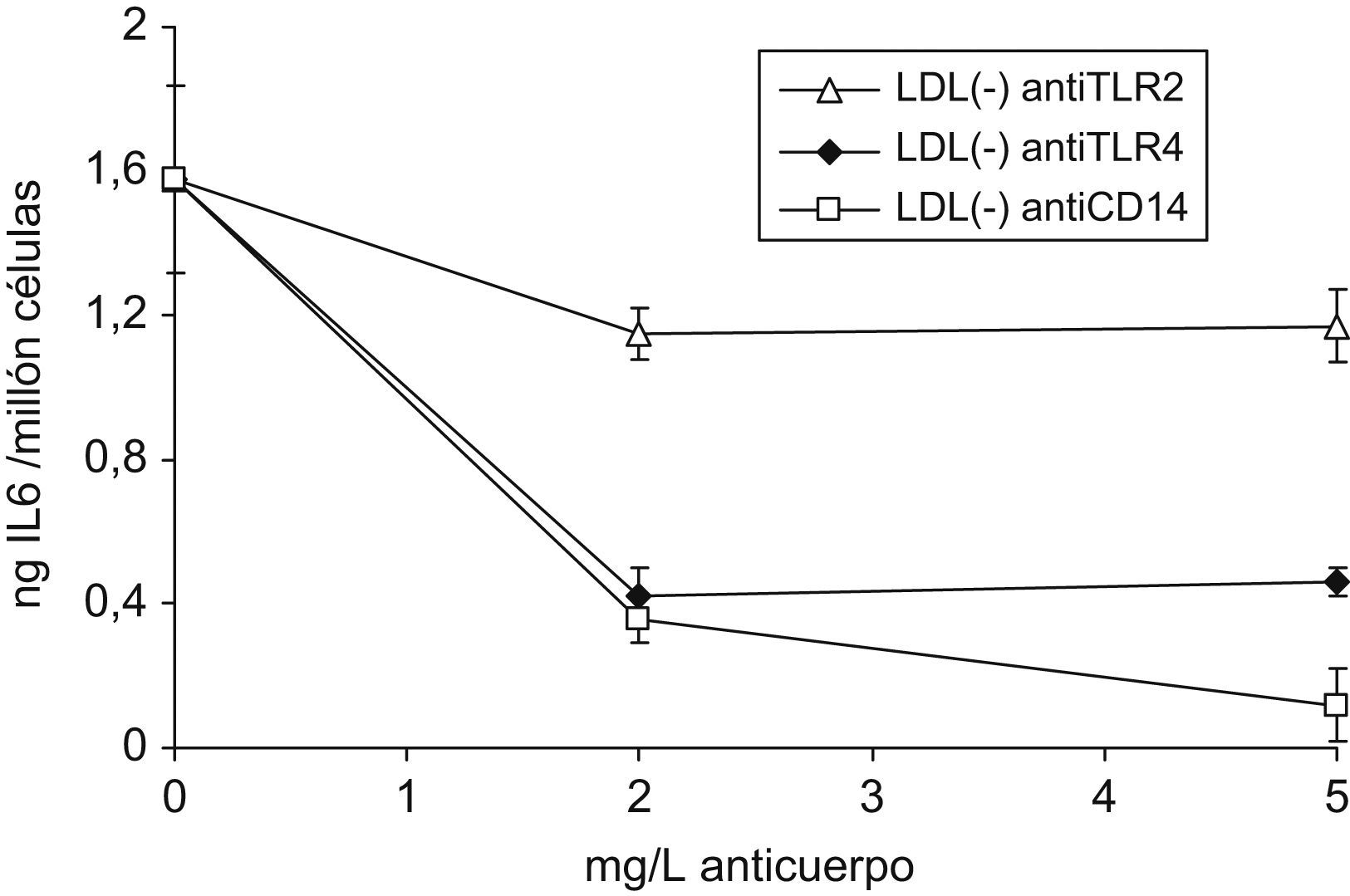
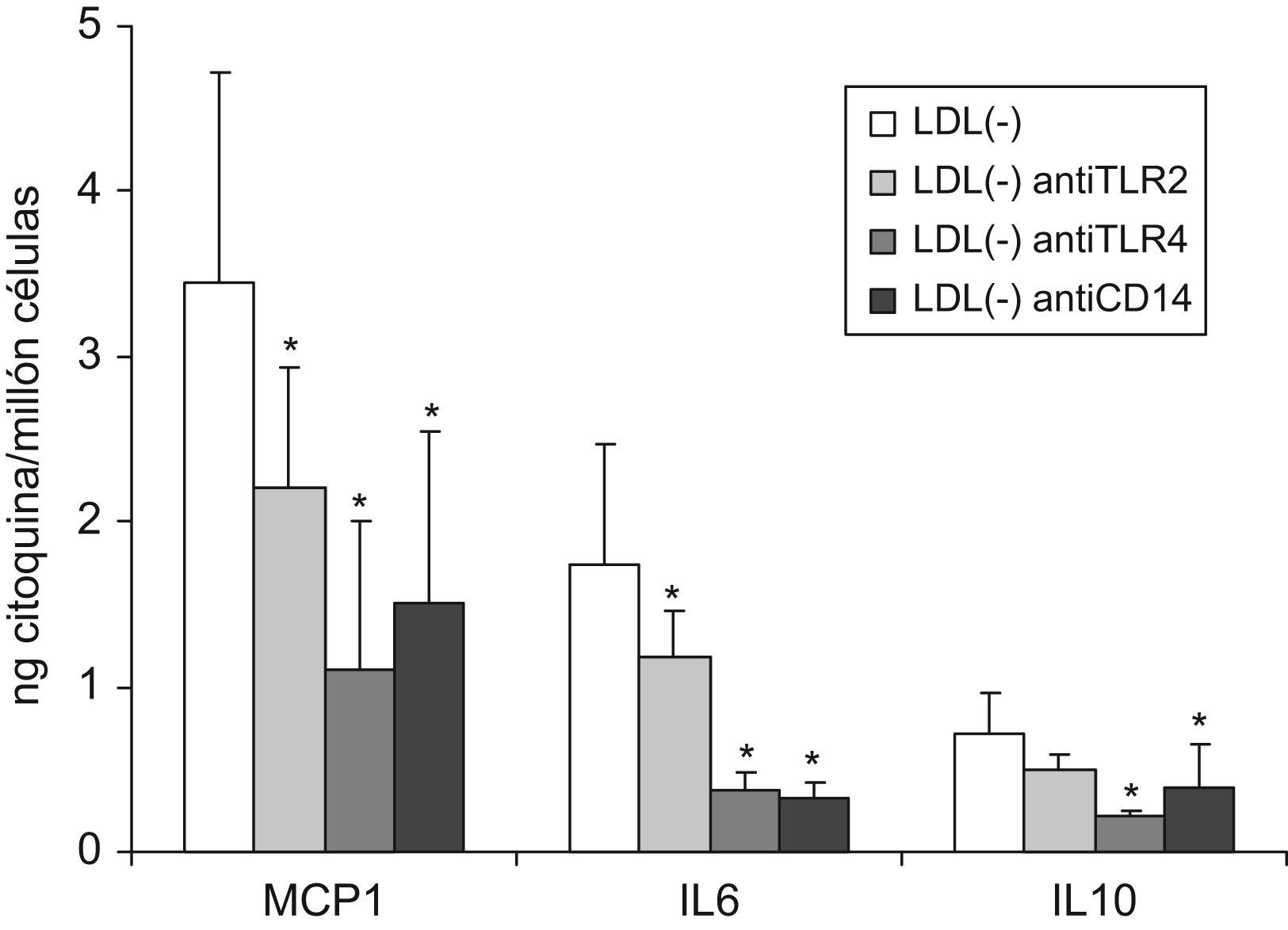
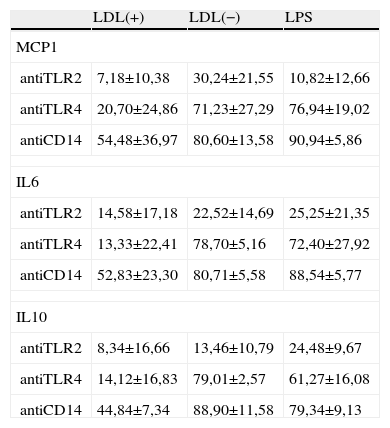
Se probaron las concentraciones de 2 y 5mg/l de los anticuerpos antiCD14, antiTLR2 y antiTLR4, observándose que en el caso del antiCD14 con la concentración más alta de anticuerpo se obtenía una mayor inhibición en la liberación de citoquinas inducida por LDL(−). Sin embargo, con antiTLR2 y antiTLR4 ya parece llegar a un máximo de inhibición con 2mg/l y no aumenta el efecto a 5mg/l. En la figura 6 se muestran los resultados en la liberación de IL6, como representativo de lo obtenido en las 3 citoquinas evaluadas. Debido a estos resultados, en estudios posteriores se usaron las concentraciones de 2mg/l para los TLRs y 5mg/l para CD14. El efecto de los anticuerpos a estas concentraciones en la liberación de MCP1, IL6 e IL10 respecto a la condición de LDL(−) sola queda reflejado en la figura 7, observándose inhibición en las 3 citoquinas por adición de los anticuerpos, excepto en la de IL10 cuando se usó el anticuerpo antiTLR2. En la tabla 2 se muestra el efecto de los mismos anticuerpos, expresado como % de inhibición en la liberación de citoquinas respecto a la condición en que no se añadió anticuerpo, es decir respecto al estímulo solo, además de LDL(−) en este caso se muestran también los resultados de LDL(+) y LPS.
. Los monocitos se preincubaron 1h con antiTLR2, antiTLR4 y antiCD14 (2 y 5mg/l) previo a la adición de LDL(−) (150mg apoB/l) durante 20h y tras la incubación se recogió el sobrenadante celular para valorar las citoquinas mediante ELISA. Se muestran los resultados de IL6 como citoquina representativa a las concentraciones indicadas de anticuerpos (triángulos: antiTLR2, rombos: antiTLR4 y cuadros: antiCD14).")
Liberación de IL6 en monocitos tratados con concentraciones crecientes de anticuerpos, previamente a la adición de LDL(−). Los monocitos se preincubaron 1h con antiTLR2, antiTLR4 y antiCD14 (2 y 5mg/l) previo a la adición de LDL(−) (150mg apoB/l) durante 20h y tras la incubación se recogió el sobrenadante celular para valorar las citoquinas mediante ELISA. Se muestran los resultados de IL6 como citoquina representativa a las concentraciones indicadas de anticuerpos (triángulos: antiTLR2, rombos: antiTLR4 y cuadros: antiCD14).
. Los monocitos se preincubaron 1h con antiTLR2 y antiTLR4 (2mg/l) y antiCD14 (5mg/l) previo a la adición de LDL(−) (150mg apoB/l) durante 20h y tras la incubación se recogió el sobrenadante celular para valorar las citoquinas mediante ELISA. Los resultados se expresan como ng de la citoquina correspondiente liberada por cada millón de células, en monocitos sin preincubar (barras blancas) o con preincubación con antiTLR2 (gris blanco), antiTLR4 (gris oscuro) o con antiCD14 (negro). Se presenta la media de 4 experimentos y se indican las significaciones estadísticas con * versus la liberación inducida por la LDL(−) en monocitos no preincubados con anticuerpos con P=0,068.")
Liberación de MCP1, IL6 e IL10 en monocitos tratados con antiTLR2, antiTLR4 y antiCD14, previamente a la adición de LDL(−). Los monocitos se preincubaron 1h con antiTLR2 y antiTLR4 (2mg/l) y antiCD14 (5mg/l) previo a la adición de LDL(−) (150mg apoB/l) durante 20h y tras la incubación se recogió el sobrenadante celular para valorar las citoquinas mediante ELISA. Los resultados se expresan como ng de la citoquina correspondiente liberada por cada millón de células, en monocitos sin preincubar (barras blancas) o con preincubación con antiTLR2 (gris blanco), antiTLR4 (gris oscuro) o con antiCD14 (negro). Se presenta la media de 4 experimentos y se indican las significaciones estadísticas con * versus la liberación inducida por la LDL(−) en monocitos no preincubados con anticuerpos con P=0,068.
Inhibición en la liberación de citoquinas inducida por LDL(+), LDL(–) o LPS por adición de antiTLR4 (2mg/l), antiTLR2 (2mg/l) o antiCD14 (5mg/l), expresada en % de inhibición respecto a la condición en que no se trataron los monocitos con anticuerpos
| LDL(+) | LDL(−) | LPS | |
| MCP1 | |||
| antiTLR2 | 7,18±10,38 | 30,24±21,55 | 10,82±12,66 |
| antiTLR4 | 20,70±24,86 | 71,23±27,29 | 76,94±19,02 |
| antiCD14 | 54,48±36,97 | 80,60±13,58 | 90,94±5,86 |
| IL6 | |||
| antiTLR2 | 14,58±17,18 | 22,52±14,69 | 25,25±21,35 |
| antiTLR4 | 13,33±22,41 | 78,70±5,16 | 72,40±27,92 |
| antiCD14 | 52,83±23,30 | 80,71±5,58 | 88,54±5,77 |
| IL10 | |||
| antiTLR2 | 8,34±16,66 | 13,46±10,79 | 24,48±9,67 |
| antiTLR4 | 14,12±16,83 | 79,01±2,57 | 61,27±16,08 |
| antiCD14 | 44,84±7,34 | 88,90±11,58 | 79,34±9,13 |
En resumen, el pretratamiento de las células con anticuerpos anti TLR2, TLR4 y CD14 inhibió la acción liberadora de citoquinas de las LDLs, pero siendo siempre superior en el caso de LDL(−) respecto LDL(+) en todas las citoquinas evaluadas, independientemente del tiempo de incubación. También inhibieron el efecto del LPS, que fue usado como control.
Comparando el efecto de los diferentes anticuerpos, se observa que la inhibición de citoquinas por pretratamiento de los monocitos con antiTLR2 previo a la adición de LDL(−) no superó el 30%, tampoco provocó una mayor inhibición sobre el efecto inflamatorio de LPS y aún menor en el caso de la LDL(+). Los efectos de antiCD14 y antiTLR4 fueron similares entre ellos en la inhibición del efecto de LDL(−), en el caso de LDL(+) y LPS se inhibió más el efecto por el bloqueo de CD14.
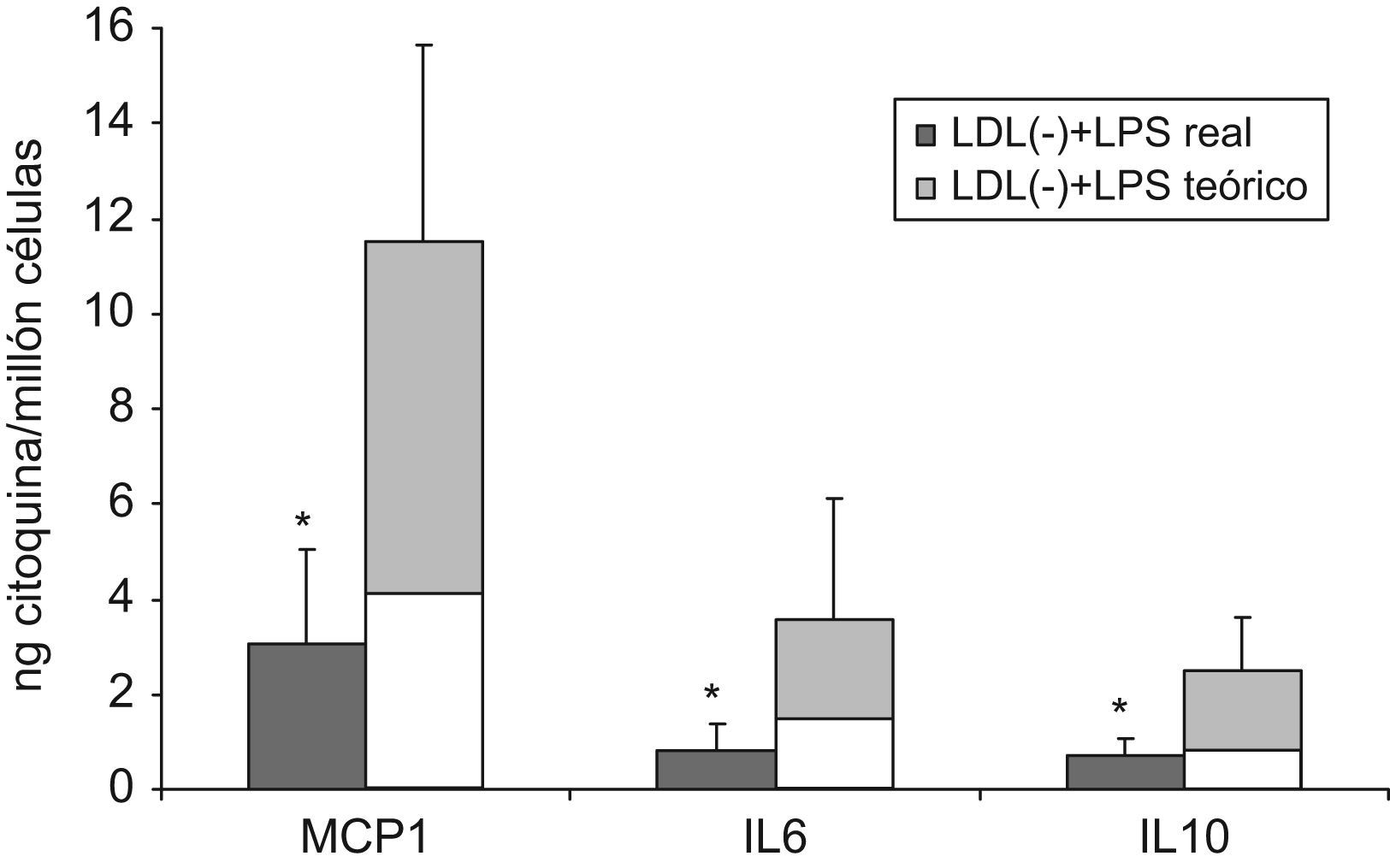
Efecto de la incubación de LDL(−) con LPS en la liberación de citoquinasEl LPS es captado principalmente por la vía TLR-CD14, por ello se quiso comprobar si la LDL(−) competía con el LPS en el aspecto de la inducción de citoquinas, o bien se sumaban sus efectos. En los estudios de captación celular ya habíamos observado una competencia por la unión a monocitos, y en la figura 8 se observa que también parece haber una competencia a nivel del efecto inflamatorio. La suma de los efectos de incubar LDL(−) con LPS en monocitos es significativamente inferior a la suma del efecto de cada estímulo por separado, tanto en MCP1 como IL6 e IL10, independientemente del tiempo de incubación de los estímulos (4 o 20h).
 y LPS, o con los dos estímulos por separado. Los monocitos se incubaron con LDL(−) (150mg apoB/l) o LPS (0,1mg/l) o ambos estímulos conjuntamente durante 20h. Tras la incubación se recogió el sobrenadante celular para valorar MCP1, IL6 e IL10. Se presentan los resultados de la liberación de citoquinas (n=4) cuando se incubaron los monocitos con LDL(−) y LPS simultáneamente (barras gris oscuro) y a la derecha la suma teórica de los dos estímulos, se indica en bloques superpuestos los valores con LPS solo (blanco) y LDL(−) sola (gris claro). Se indican las significaciones estadísticas con * versus la liberación teórica consecuencia de la suma de los efectos de LDL(−) y LPS con P=0,068.")
Liberación de MCP1, IL6 e IL10 en monocitos tratados simultáneamente con LDL(−) y LPS, o con los dos estímulos por separado. Los monocitos se incubaron con LDL(−) (150mg apoB/l) o LPS (0,1mg/l) o ambos estímulos conjuntamente durante 20h. Tras la incubación se recogió el sobrenadante celular para valorar MCP1, IL6 e IL10. Se presentan los resultados de la liberación de citoquinas (n=4) cuando se incubaron los monocitos con LDL(−) y LPS simultáneamente (barras gris oscuro) y a la derecha la suma teórica de los dos estímulos, se indica en bloques superpuestos los valores con LPS solo (blanco) y LDL(−) sola (gris claro). Se indican las significaciones estadísticas con * versus la liberación teórica consecuencia de la suma de los efectos de LDL(−) y LPS con P=0,068.
La LDL electronegativa (LDL(−)) es una subfracción plasmática de LDL cuya proporción está aumentada en patologías con elevado riesgo cardiovascular. Uno de los principales efectos descritos para la LDL(−) es la inducción de diferentes moléculas inflamatorias en células endoteliales y células mononucleares circulantes. Estos datos sugieren que la LDL(−) podría constituir un importante factor aterogénico; sin embargo, existe un gran desconocimiento respecto a las vías que median su efecto. Este estudio se ha centrado en los receptores celulares implicados en la captación de la LDL(−), la cual desencadena la inducción de citoquinas en monocitos humanos.
En el presente trabajo se obtuvo que, al igual que en células endoteliales8, la LDL(−) se une a los monocitos cuantitativamente de igual manera que la LDL(+). Estos datos no evaluan la afinidad por los distintos receptores, por tanto el que no hayan diferencias respecto a LDL(+) no descarta que la unión sea más o menos débil, que exista unión inespecífica o diferencias en captación por algunos receptores determinados. Este último aspecto es el que se ha tratado en este estudio, mediante el abordaje de bloquear diferentes receptores candidatos y comprobar si disminuía la unión total de la LDL(−) a monocitos y esto se podía traducir en una disminución de su efecto liberador de citoquinas. Concretamente, como citoquinas representativas de las inducidas por LDL(−) se evaluaron la quimioquina atrayente de monocitos MCP1, la citoquina inflamatoria de efecto pleiotrópico IL6 y la interleuquina reguladora con propiedades antiinflamatorias IL10.
No se obtuvo una mayor liberación de citoquinas tras cultivar las células en medio deficiente para sobrexpresar el rLDL (datos no mostrados). Esto descarta que la entrada por esta vía fuera la que promueve la inducción de citoquinas, lo cual coincidiría con el hecho de que se sabe que la LDL(−) presenta menor afinidad por el rLDL2 y que además los monocitos expresan escasamente este receptor.
Los PG pueden unir lipoproteínas y estos complejos internalizarse en las células, como es el caso de la captación de LDL oxidada por PG de CS24. Como nuestro grupo ha descrito que la LDL(−) se une con mayor afinidad que la LDL(+) a los proteoglicanos, sobre todo a los de condroitín sulfato4, se planteó que esta podría ser una vía de entrada de LDL(–). En monocitos el PG más abundante es el de condrotín sulfato25 por ello se utilizó condroitinasa para eliminarlo, sin embargo, ni esto ni el uso de condrotín sulfato como competidor para unir LDL(−), no alteró la unión total de LDL(−) marcada a los monocitos, por lo que no parece ser una vía importante de entrada a la célula. Tampoco se observaron diferencias en la condición en que los monocitos fueron diferenciados con PMA para hiperexpresar PG. Estos resultados no descartan que de forma minoritaria la LDL(−) pueda entrar vía PG e inducir la liberación de citoquinas, hecho que parece cierto en el caso de MCP1, en el que al degradar los PG celulares hubo una inhibición de su liberación, especialmente tras 20h de incubación. Respecto al aumento en la liberación de citoquinas tras tratamiento con condroitinasa en la condición basal sería debido a una menor retención de las mismas en la superficie celular a través de los PG o al hecho de que se ha descrito que los GAG libres son inflamatorios26.
Por otra parte, se conocía que la LDL(−) no era captada por SRAII2, pero existen muchos otros tipos de SR que podrían interaccionar con LDL(−) y mediar su efecto a nivel celular. Se ha descrito que LOX capta la LDL(−) en células endoteliales14, pero este receptor está poco expresado en el caso de los monocitos y estudios previos de expresión génica de nuestro grupo también indican que los monocitos que aislamos tienen una baja expresión de este receptor10. En cambio, sí hay expresión de CD36, que es otro SR ampliamente estudiado que puede unir LDL modificadas16. Los resultados indican que el uso de anticuerpos anti-CD36 no afectó a la unión total de LDL(+) ni LDL(−) a los monocitos, por lo que tampoco parece una entrada importante de LDL(−) a nivel celular. En cambio, el bloqueo de este receptor provocó una disminución en la liberación de IL6 por LDL(−), aunque no de MCP1 ni IL10. Estos datos parecen indicar que las diferentes citoquinas producidas por acción de la LDL(−) pueden inducirse por diferentes vías, teniendo el CD36 algún papel en la liberación de IL6 y los PG en la de MCP1.
En nuestros experimentos de liberación de citoquinas siempre se usó LPS como control positivo, ya que induce una respuesta similar/superior a la LDL(−). Se conoce que el LPS, además de ser un estímulo por sí solo, potencia los efectos de otros inductores como TNF, IL1 o LDL oxidada; se ha descrito que ésta última no induce citoquinas proinflamatorias en monocitos humanos si no hay coestimulación con LPS27. Sin embargo, al incubar los monocitos con LPS y LDL(−) no se produjo un efecto sinérgico, ni siquiera aditivo, sinó que la liberación de citoquinas fue significativamente inferior a la esperada. Esta disminución en la liberación de citoquinas coincubando los monocitos con LDL(–) y LPS podría ser por competencia entre ellos por vías comunes que conlleven a la inducción de citoquinas o bien por neutralización del LPS. En este último aspecto, se ha descrito que la preincubación de lipoproteínas, y especialmente LDL, con LPS neutraliza la inducción de citoquinas inducida por el mismo en células mononucleares28–30. Por otra parte, se ha descrito que diferentes miembros de la familia del receptor de LPS están en la membrana celular en dominios ricos en esfingomielina y estímulos como los fosfolípidos oxidados aumentan la actividad SMasa, inhibiendo la acción del LPS31. También un aumento en la actividad fosfolipasa C (PLC) elimina CD14 y disminuye la captación de LPS28. Esta neutralización del efecto del LPS podría ser factible con la LDL(−) que ya de por sí presenta estas actividades fosfolipolíticas aumentadas5. Sin embargo, nuestros resultados indican que entre LDL(−) y LPS lo que más probablemente existiría sería compartición de vías de inducción de citoquinas y, por tanto, al ponerlos simultaneamente se produciría una competición entre ambos y no se sumarían sus efectos. Concretamente, habría una competición en la captación celular por los receptores, según los estudios de desplazamiento. Además, otros resultados que apoyan esta hipótesis son que el bloqueo de receptores implicados en la unión y efecto de LPS en monocitos (TLR2, TLR4 y CD14) provocó inhibición de la liberación de citoquinas por parte de la LDL(−).
Por otro lado, se ha descrito que los fosfolípidos oxidados pueden inhibir la acción inflamatoria del LPS por competición con TLR2 y TLR432 o con CD1433. Miller et al propuso que la LDL mínimamente oxidada (LDLmm) compartía la misma vía que LPS en la inducción de citoquinas, aunque descartaba el TLR234 y posteriormente describió que podía ser por la vía de TLR4 aunque también existía otra vía independiente de TLR419. La unión de LDLmm a TLRs podía ser directa o a través de su reconocimiento por CD1419, mientras que ni la LDL nativa ni la extensamente oxidada tienen la capacidad de unirse a CD1434. Está descrito que CD14 une ceramida35 y ácidos grasos36, ambos componentes aumentados en la LDL(−), por lo que la LDL(−) podría unirse más a CD14 que la LDL(+) debido a ello.
Se ha descrito también que el LPS activa la vía de la ceramida37 y, por otra parte, que la activación de CD14 induce una actividad tipo PLC que genera ceramida, promoviendo activación de TLR4 y producción de citoquinas38. Por ello, se puede hipotetizar que la propia actividad PLC de la LDL(−) conllevaría a una mayor producción de ceramida añadida a la producida por la activación de CD14, y esto podría ser responsable del mayor efecto de liberación de citoquinas de LDL(−) respecto a la LDL nativa.
El CD14 está implicado en la presentación de ligandos a los TLRs, que son receptores menos abundantes, aunque si la concentración del ligando es alta sí se puede unir directamente a TLR independiente de CD1437. Los resultados obtenidos muestran que el bloqueo de CD14 hizo disminuir la unión a monocitos de la LDL(−), pero también el bloqueo de TLR2 y TLR4 produjo este efecto. Esto mismo se observó en la liberación de citoquinas, donde el uso de antiCD14 fue la forma más eficaz de disminuir la producción de citoquinas inducida por la LDL(−), aunque se obtuvo un efecto similar con TLR4 y en menor grado con TLR2. De todas formas tanto TLR2 como TLR4 se asocian a CD14 por lo no se puede tener la certeza de que al bloquear uno de los miembros del complejo se impida la unión a otro de los componentes.
En cualquier caso, los resultados indican que la competición que se observó por parte de la LDL(−) con el efecto del LPS es debida a que comparten vías comunes de activación de citoquinas. El LPS es capaz de desplazar la unión de la LDL(−) con gran eficacia, lo cual indicaría que presenta mayor afinidad por los receptores por los que compiten, presumiblemente CD14 o TLRs, pero hay un porcentaje de unión de LDL(−) que no puede inhibirse aunque se aumente la concentración de LPS, la cual podría ser unión a otros receptores no compartidos con el LPS.
La competición de la LDL(−) por las vías de activación de LPS podría sugerir un efecto protector de la LDL(−) en situaciones inflamatorias en que hubiera una concentración de LPS elevada. Pero también se ha descrito en el caso de los fosfolipídos oxidados que su competición con LPS impediría la fagocitosis en caso de infección bacteriana39. En conjunto, hay que considerar que el efecto global dependerá del balance de varios factores entre ellos la concentración de LPS y de LDL(−).
En resumen, la vía de TLR2 y TLR4 junto con CD14, por la que competiría con LPS, parece ser importante en el desencadenamiento de los efectos inflamatorios de la LDL(−) en monocitos. Por otra parte, los resultados indican que en las citoquinas valoradas también existen otras vías de inducción y que la LDL(−) podría actuar por diferentes mecanismos. Por lo tanto, sería de gran interés profundizar en el papel de estas vías y evaluar otros posibles mecanismos de entrada de la LDL(−) a las células.
FinanciaciónEste trabajo ha sido parcialmente financiado por las ayudas al proyecto de investigación básica de la Sociedad Española de Arteriosclerosis (SEA) 2007 y al proyecto FIS CP04-0110.