






Introducción
La prevalencia del síndrome metabólico está incrementándose a nivel mundial1, con un aumento paralelo del riesgo de desarrollar diabetes mellitus y enfermedad cardiovascular, así como de la mortalidad asociada a estas enfermedades2. Aunque es poco factible que exista una causa única de esta tendencia, factores ambientales (dieta, actividad física, estrés, etc.), junto con factores genéticos de predisposición se encuentran probablemente involucrados. Paralelamente al incremento en el consumo de energía total producido durante las últimas décadas, se ha producido también en todo el mundo un cambio en el tipo de nutrientes consumidos, con un consumo aumentado de fructosa, sobre todo debido a la mayor ingesta de refrescos y otros tipos de bebidas con un alto contenido en fructosa3. Una vez absorbida, la fructosa dietética es captada mayoritariamente por el hígado, en donde promueve la síntesis de triglicéridos y la producción de lipoproteínas de muy baja densidad (VLDL). Sin embargo, la modificación en la síntesis de ácidos grasos no explica en su totalidad la elevada producción de triglicéridos4. En consecuencia, no conocemos en su totalidad los mecanismos que causan la alteración en el metabolismo lipídico hepático producida por dietas ricas en fructosa.
La rata es un buen modelo para estudiar el metabolismo humano de la fructosa5. Una dieta rica en fructosa induce en la rata alteraciones metabólicas similares a las encontradas en el síndrome metabólico, por lo que la rata alimentada con fructosa es un modelo aceptado de síndrome metabólico. Sin embargo, mientras que los diseños experimentales que utilizan proporciones de fructosa del 50% o superiores en dietas sólidas inducen hipertrigliceridemia y una marcada resistencia a la insulina6,7, las dietas que incorporan fructosa al 10% (peso/volumen) en el agua de bebida por períodos cortos inducen hipertrigliceridemia e hígado graso, sin modificar los valores plasmáticos de glucosa e insulina8,9. Dado que es bien conocido que tanto los valores elevados de hidratos de carbono como la hiperinsulinemia crónica incrementan la síntesis lipídica hepática y la producción de VLDL10, escogimos la segunda aproximación dietética (fructosa al 10% peso/volumen [p/v] en el agua de bebida) para examinar los efectos de la fructosa en el metabolismo hepático de los ácidos grasos sin la interferencia producida por los efectos de la hiperinsulinemia.
En estudios previos utilizando este mismo modelo experimental hemos demostrado que la administración de fructosa a ratas produce una marcada reducción en la betaoxidación hepática de ácidos grasos y en los valores de ARN mensajero (ARNm) para el receptor activado por proliferadores peroxisómicos * (PPAR*)9. En el presente trabajo, demostramos que la administración de fructosa al 10% (p/v) en el agua de bebida de ratas reduce la actividad de transactivación y transrepresión de PPAR*, sin que se afecten los valores hepáticos de proteína PPAR*. Como consecuencia, la fructosa deprime la oxidación hepática de ácidos grasos e incrementa la actividad del factor de transcripción proinflamatorio NF-*B. Estos cambios no se observaron en ratas a las que se administró glucosa al 10% (p/v) en el agua de bebida. Dado que la ingestión de ambos hidratos de carbono produjo cambios similares en la expresión hepática de factores de trascripción y enzimas implicados en la síntesis de ácidos grasos, concluimos que la hipertrigliceridemia y la esteatosis hepática inducidas por la ingestión de fructosa son consecuencia de la reducción en el catabolismo de ácidos grasos.
Material y métodos
Animales y diseño experimental
Se utilizaron ratas macho Sprague-Dawley (Criffa, Barcelona) que se mantuvieron durante 5 días con agua y comida ad libitum, a temperatura y humedad constante, con un ciclo de luz/oscuridad de 12 h. Tras el período de aclimatación, los animales se distribuyeron aleatoriamente en 3 grupos de 8 animales cada uno: un grupo control, un grupo suplementado con fructosa (fructosa) y un grupo suplementado con glucosa (glucosa). Ambos hidratos de carbono se administraron en el agua de bebida en forma de solución al 10% (peso/volumen) durante 2 semanas. Los animales control no recibieron ningún suplemento de hidratos de carbono. Se sacrificó a los animales por decapitación bajo anestesia por ureatano entre las 9-10 horas. Todo el proceso se realizó de acuerdo con la guía establecida por el Comité de Bioética de la Universidad de Barcelona, según se indica en la Ley (5/1995) (21 de julio) de la Generalitat de Catalunya.
Preparación de muestras
Las muestras sanguíneas se recolectaron en el momento del sacrificio en tubos con EDTA al 5%; el plasma se obtuvo por centrifugación y se conservó a 80 ºC hasta su utilización. Los hígados de las ratas se extrajeron y perfundieron en tampón 150 mM NaCl, 1 mM DTT, 30 mM EDTA, 50 mM KH2PO4, pH 7,4; 0,5 g de tejido hepático de cada rata se homogenizó en el mismo tampón para obtener la fracción de sobrenadante posnuclear, que se conservó a 80 ºC hasta su utilización; se congeló 10-100 mg de tejido hepático en N2 líquido de forma inmediata y conservados a 80 ºC, hasta su utilización para la extracción del ARN total. Dos muestras adicionales de tejido hepático (250 mg) se conservaron a 80 ºC para la cuantificación de los lípidos hepáticos y para la obtención de los extractos nucleares y de proteína total.
Análisis de lípidos, glucosa, insulina y leptina
Las concentraciones plasmáticas de triglicéridos, ácidos grasos libres (AGL) y glucosa se midieron mediante los ensayos colorimétricos de Wako Chemicals GmbH (Neuss, West Germany) Triglyceride L-Type, NEFAC y Glucose kit, respectivamente. La concentración plasmática de insulina y de leptina se determinó por radioinmunoanálisis (RIA) usando los kits comerciales RPA547 de Amersham Biosciences Europe GMBH y RL83K de Linco Research (Missouri, USA), respectivamente. Los lípidos hepáticos se extrajeron y midieron según se describe previamente9.
Ensayos enzimáticos
La actividad hepática de betaoxidación de ácidos grasos se determinó según el método descrito previamente11, utilizando 30 µg del sobrenadante posnuclear como muestra.
Preparación y análisis del ARN
El ARN total se aisló mediante el reactivo Ultraspec (Biotecx, Houston, USA), de acuerdo con las instrucciones del fabricante. Los valores relativos de cada ARN específico se determinaron por la reacción de la transcriptasa reversa-reacción en cadena de la polimerasa (RT-PCR), tal como se ha descrito previamente9. El ADNc monohebra se sintetizó a partir de 1 µg de ARN total hepático, utilizando como cebadores 125 ng de hexámeros aleatorios y 200 U M-MLV-Reverse-Transcriptase (RT) en un tampón 50 mM Tris-HCl (pH 8,3), con 75 mM KCl, 3 mM MgCl2, 10 mM DTT, 20 U RnaseOut y 500 µM de cada dNTP en un volumen total de 20 µl. La reacción de RT se realizó durante 60 min a 37 ºC. La reacción de PCR se realizó utilizando una alícuota de 5 µl de la mezcla de reacción de RT, 0,5 µg de cada uno de los cebadores sentido y antisentido, 200 µM dNTPs, 1 U Taq DNA polimerasa y 1,25 µCi a-[32P]-dATP en 20 mM Tris-HCl, pH 8,5, 2,5 mM Mg Cl2 (volumen final de 50 µl). Para evitar anillamientos inespecíficos, el ADNc y la Taq polimerasa los dNTPs se separaron de los cebadores mediante un tapón de parafina. En la fase de desnaturalización inicial, la parafina funde a 60 ºC, lo que permite la mezcla de los componentes de la reacción. La reacción de PCR se realizó en un aparato MJ Research Thermocycler equipado con un sistema Peltier y sonda de temperatura. Después de la desnaturalización de los cebadores y el ADNc a 94 ºC durante 1 min, el programa cíclico se realizó como sigue: 92 ºC durante 1 min, 60 ºC (63 ºC para SREBP-1C) durante 1 min y 15 s y 72 ºC durante 1 min y 50 s. En el último ciclo, se realizó una extensión final de 5 min a 72 ºC. Para confirmar la ausencia de contaminación, en cada experimento se incluyeron controles negativos. Se han realizado experimentos preliminares para determinar las condiciones de amplificación exponencial para todos los genes estudiados, calculando el rango de número de ciclos entre los cuales se produce una relación linear entre el ARN incorporado y el producto final. Para el conjunto de cebadores, se realizó una progresión de ciclos de PCR (con el resto de las condiciones fijas), a fin de determinar el número óptimo de ciclos. El mismo procedimiento se siguió para determinar las concentraciones de ARN12. Se utilizó la adenosina fosforribosil transferasa (APRT) como control interno, coamplificando con las secuencias valoradas en el mismo tubo, excepto en el caso de la ácido graso sintasa (FAS), estearoil-CoA desaturasa-1 (SCD1), piruvato cinasa hepática (LPK) y la glucosa-6-fosfatasa (G6PC). Estas secuencias se amplificaron en paralelo con APRT, en duplicado y en tubos separados. El número de ciclos, las secuencias de los cebadores y los productos resultantes de PCR se muestran en la tabla 1. Se sometieron 5 µl de cada mezcla de reacción de PCR a electroforesis al 5% en gel de poliacrilamida en 1*TBE. Los geles se secaron, se autorradiografiaron en película RX-OMAT S Kodak, y se cuantificaron por rastreo vídeo-densitométrico (programa IMAT program, Servicios Científico Técnicos, Universidad de Bar celona). Los valores de ARNm siempre se expresaron en función de su relación con los valores de ARNm para APRT.

Aislamiento de los extractos nucleares y de la proteína total
Los extractos nucleares se aislaron por el método de Helenius13, con algunas modificaciones. Brevemente, los tejidos hepáticos se pesaron y homogenizaron en un homogenizador Potter Elvehjem en 4 volúmenes (p/v) de tampón hipotónico que contenía 0,25 M sacarosa, 10 mM Hepes, pH 7,9, 10 mM KCl, 1,5 mM MgCl2, y una mezcla de inhibidores de proteasas y fosfatasas (0,2 mM fluoruro de fenilmetilsulfonilo, 0,5 mM ditiotreitol, 5 µg/ml aprotinina, 2 */ml leupeptina, 100 mM NaF y 1 mM ortovanadato). Los homogenizados se incubaron en hielo 10 min y se centrifugaron a 25.000 * g durante 15 min. Los pellets se lavaron con 4V del mismo tampón hipotónico y se centrifugaron a 10.000 * g durante 10 min. Los pellets resultantes se resuspendieron con 2 volúmenes de tampón 20 mM Hepes, pH 7,9, 1,5 mM MgCl2, 20 mM KCl, 0,2 mM EDTA, 25% glicerol que contenía los mismos inhibidores de proteasas y fosfatasas antes mencionados. Posteriormente, se añadió gota a gota un volumen de tampón rico en sales (20 mM Hepes, pH 7,9, 1,5 mM MgCl2, 1,2 M KCl, 0,2 mM EDTA, 25% glicerol e inhibidores de proteasas y fosfatasas) y se mantuvo la suspensión en agitación durante 30 min. Tras centrifugación a 25.000 * g durante 30 min. Se guardaron los sobrenadantes a 80 ºC hasta su utilización. Todo el proceso se llevó a cabo a 4 ºC. La proteína posnuclear se obtuvo por homogeneización de 200 mg de tejido hepático en un tampón 20 mM Tris-HCl, pH 7,5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 100 mM NaF, 1% Igepal, 0,1 µg/ml ácido ocadaico, 1 mM ortovanadato, 1 mM PMSF y 2 µg/ml leupeptina. Los homogenizados se mantuvieron 30 min a 4 ºC y se centrifugaron a 15.000 * g durante 15 min a 4 ºC. Los sobrenadantes se guardaron en alícuotas a 80 ºC. La cuantificación de la concentración proteica de cada fracción se realizó mediante el método de Bradford14.
Ensayo de retardación de la movilidad electroforética (EMSA)
Las secuencias de los oligonucleótidos de doble cadena utilizadas fueron las siguientes: oligonucleótido concensus del factor nuclear *B (NF-*B) 5'-AGTTGAGGGGACTTTCCCAGGC-3' (Promega, Madison, USA); elemento de respuesta a PPAR (PPRE situado en la posición 266 /290 del promotor del gen de la L-CPTI) 5'-AGTACGGGCATGGAGCAAAGAGCT-3'. Se marcaron radiactivamente los oligonucleótidos mediante la siguiente reacción: 2 µl de cada oligonucleótido (1,75 pmol/µl), 2 µl del tampón cinasa 5, 5 U de la enzima T4 polinucleótido cinasa y 3 µl de [*-32P] ATP (3000 Ci/mmol a 10 mCi/ml, Amersham) incubado a 37 ºC durante 2 h. La reacción se paró añadiendo 90 µl de tampón TE (10 mM Tris-HCl, pH 7,4 y 1 mM EDTA). Para separar la sonda marcada del ATP no unido se eluyó la mezcla de reacción en una columna Nick (Pharmacia, Sant Cugat, España) siguiendo las instrucciones del fabricante; 8 µg de extracto nuclear se incubaron durante 10 min en hielo con el tampón de unión (10 mM Tris-HCl, pH 8,0, 25 mM KCl, 0,5 mM DTT, 0,1 mM EDTA, pH 8,0, 5% glicerol, 5 mg/ml BSA y 50 µg/ml poly(dI-dC)) en un volumen final de 15 µl. La sonda marcada (aproximadamente 60.000 cpm) se añadió a la reacción y se incubó 20 min a temperatura ambiente (NF-*B a 4 ºC). En los ensayos de competición se incubó 15 min en hielo con el oligonucleótido específico no marcado antes de añadir la sonda marcada radiactivamente. En los ensayos de superretardación, los anticuerpos específicos se añadieron antes de la sonda marcada y se incubaron durante 30 min a 4 ºC. Los complejos proteína-ADN se sometieron a electroforesis a 4 ºC en un gel de poliacrilamida del 5% y posteriormente se realizó una autorradiografía. Los anticuerpos de PPAR*, RXR*, P65 y Oct-1 (octamer motif-1 transcription factor) se obtuvieron de Santa Cruz Biotechnology (Santa Cruz, CA).
Ensayos de Western blot
Treinta µg de sobrenadante posnuclear (para el precursor de SREBP1c) o 40 µg de extracto nuclear (para PPAR*, SREBP1c maduro y ChREBP) procedentes de hígado de rata se sometieron a electroforesis en un gel de poliacrilamida-SDS al 10% (8% para SREBP1c). Posteriormente, las proteínas se trasfirieron a una membrana Immobilon polyvinylidene diflouride (PVDF) (Millipore, a Bedford, MA) y se bloquearon durante 1 h a temperatura ambiente con un 5% de leche en polvo desnatada en TBS-0,1% Tween-20. Las membranas se incubaron con los anticuerpos primarios: anti-PPAR* (dilución 1:1000)15, anti-SREBP1 (ambas formas a dilución 1:200) y anti-ChREBP (dilución 1:500) en solución de bloqueo a 4 ºC durante toda la noche. Seguidamente, y tras varios lavados, se incubaron las membranas con los correspondientes anticuerpos secundarios (dilución 1:3000). La detección se realizó usando el kit de quimiluminiscencia ECL de Amersham Biosciences. Como control de carga se tiñeron las membranas con rojo Ponceau S16. El tamaño de las proteínas detectadas se estima gracias a los marcadores de peso molecular (Invitrogen, Life Technologies). Todos los anticuerpos son de Santa Cruz Technologies.
Coinmunoprecipitación
Cincuenta µg de extracto nuclear se incubaron con 4 µg de anticuerpo anti-p65 en un volumen final de 0,5 ml en tampón PBS (10 mM) y 2% de albúmina (BSA) durante 6 h a 4 °C. Los inmunocomplejos se obtuvieron incubando las muestras con una suspensión de proteína A-agarosa (Santa Cruz Biotechnology) toda la noche a 4 °C y en agitación constante. Los pellets de proteína A-agarosa se recogieron tras centrifugarlos y se lavaron 3 veces con PBS e inhibidores de proteasas. Tras la última centrifugación los pellets se resuspendieron con 60 µl de tampón de carga y se hirvieron a 100 ºC durante 5 min. Una alícuota del sobrenadante se sometió a electroforesis al 10% SDS y se prosiguió con el Western blot usando como anticuerpo primario el anti-PPAR*.
Estadística
Los resultados se expresan como la media ± desviación estándar. Las muestras de plasma se analizaron por duplicado. Las diferencias estadísticamente significativas se valoraron mediante la prueba de la t de Student para muestras no apareadas o la prueba de ANOVA (con análisis a posteriori), usando el programa informático GraphPad InStat (GraphPad Software V2.03). Cuando el número de muestras era pequeño o las varianzas eran no homogéneas se aplicó una prueba no paramétrica (prueba de Mann-Whitney o de Kruskal-Wallis). Se consideró significación estadística un valor de p < 0,05.
Resultados
Las ratas alimentadas con fructosa son hipertrigliceridémicas e hiperleptinémicas y muestran esteatosis hepática en comparación a las ratas alimentadas con glucosa, aun consumiendo la misma cantidad de energía. Tras 14 días de ingestión de hidratos de carbono, las ratas alimentadas con fructosa mostraron un incremento de 1,9 y 2,4 veces en los valores de triglicéridos y leptina plasmáticos, respectivamente. El contenido hepático de triglicéridos también se vio incrementado en estos animales (1,8 veces respecto a los animales control). La administración de glucosa al 10% en el agua de bebida no produjo estos incrementos. No obstante, ambos grupos de animales consumieron cantidades similares de dieta (13% menos de ingesta las ratas fructosa y el 22% menos las ratas glucosa frente a control) y de agua rica en hidratos de carbono (2,4 veces más en los dos grupos de animales frente a grupo control). Ni el peso final de los animales, ni los valores de insulina y glucosa plasmáticos se modificaron en ningún grupo de estudio; sólo la administración de glucosa provocó un incremento en los valores de AGL que fue de 1,4 veces (tabla 2).

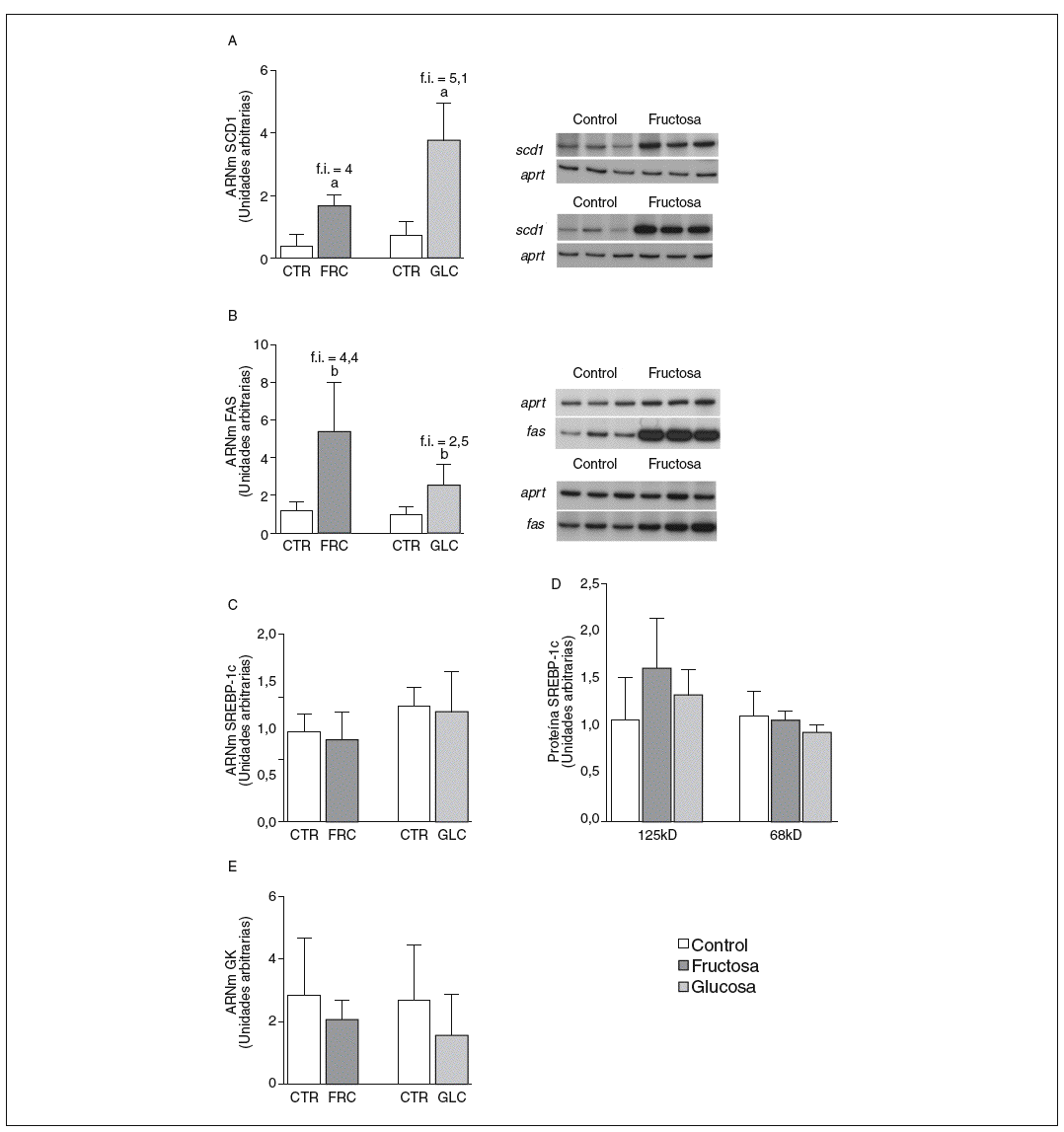
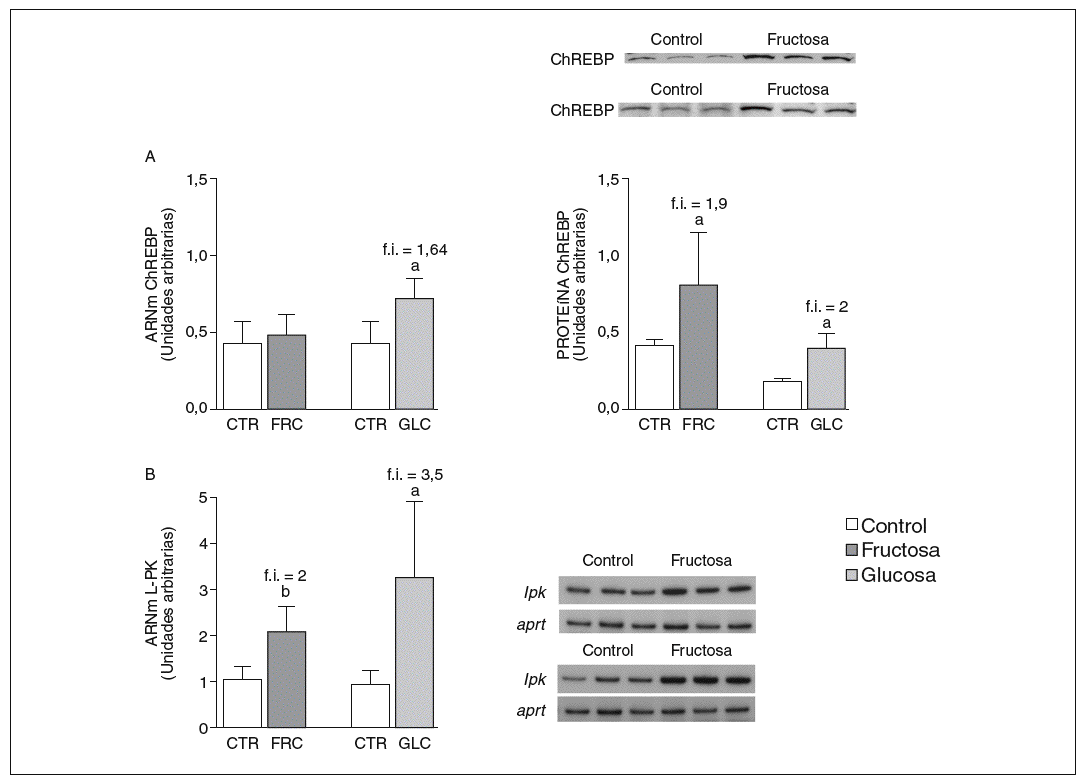
La fructosa y la glucosa producen cambios similares en factores de trascripción hepáticos que controlan la síntesis de ácidos grasos. Los valores de ARNm para SCD1, enzima que controla la biosíntesis hepática de ésteres de colesterol y triglicéridos17, se vieron incrementados 4 y 5,1 veces en los animales a los que se administró fructosa y glucosa, respectivamente, respecto a los animales del grupo control. El ARNm de FAS, enzima que regula la síntesis de ácidos grasos saturados18, se vio incrementado tanto en los animales tratados con fructosa como glucosa frente al grupo control (4,4 y 2,5 veces, respectivamente) (fig. 1A y B). Sin embargo, los valores de ARNm y proteína de SREBP1c, factor de transcripción clave en el control de la síntesis de ácidos grasos en el hígado19, no se modificaron en ningún grupo de tratamiento (fig. 1C y D). Los valores de ARNm de GK (glucocinasa), gen regulado directamente por SREBP1c20, tampoco se vieron modificados tras la ingesta de hidratos de carbono (fig. 1E). Por el contrario, la expresión de ChREBP, otro factor de transcripción implicado directamente en la regulación transcripcional de genes lipogénicos por hidratos de carbono en el hígado19, se incrementó tras la administración de fructosa y glucosa, así como los niveles de ARNm de LPK, gen regulado directamente por ChREBP21, que incrementaron 2 veces en los animales a los que se administró fructosa y 3,5 veces en los tratados con glucosa con respecto las ratas control (fig. 2A y B). La activación transcripcional de ChREBP producida por los 2 hidratos de carbono incrementó de forma similar la expresión hepática de genes lipogénicos tras el tratamiento; no obstante, sólo los animales tratados con fructosa mostraron hipertrigliceridemia y esteatosis hepática (tabla 2).
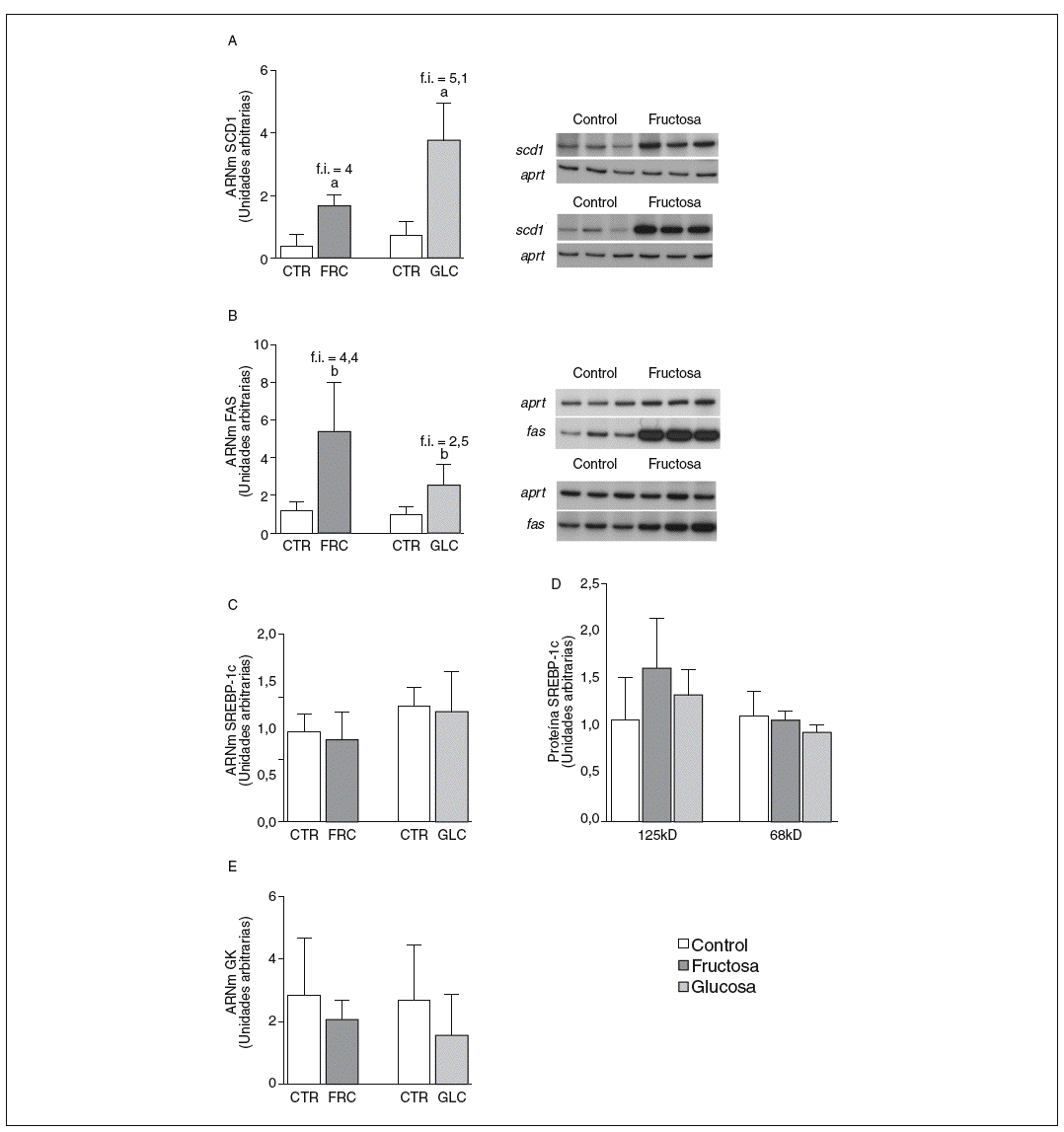
Figura 1. Niveles de expresión de enzimas clave implicadas en la síntesis de ácidos grasos y SREBP tras la administración de hidratos de carbono. A: niveles relativos de ARNm para el gen scd1 en muestras hepáticas de animales control (CTR) y animales tratados con fructosa (FRC) y con glucosa (GLC). Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. A la derecha del gráfico se muestra una autorradiografía representativa de la determinación por RT-PCR para 3 animales CTR y 3 animales tratados con FRC y GLC. B: niveles relativos de ARNm para el gen fas en muestras hepáticas de animales CTR y animales tratados con FRC y GLC. Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. A la derecha del gráfico se muestra una autorradiografía representativa de la determinación por RT-PCR para 3 animales control y 3 animales tratados con FRC y GLC. C: niveles relativos de ARNm para el gen srebp-1c en muestras hepáticas de animales CTR y animales tratados con FRC y GLC. Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. D: niveles de proteína SREBP-1c, forma precursora (125 KD) y forma madura (68 KD), en muestras hepáticas de animales CTR y animales tratados con FRC y GLC. E: niveles relativos de ARNm para el gen gk en muestras hepáticas de animales CTR y animales tratados con FRC y GLC. Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. Todos los resultados están expresados en unidades arbitrarias. ap < 0,001. bp < 0,01. cp < 0,05. f.i.: fold induction .
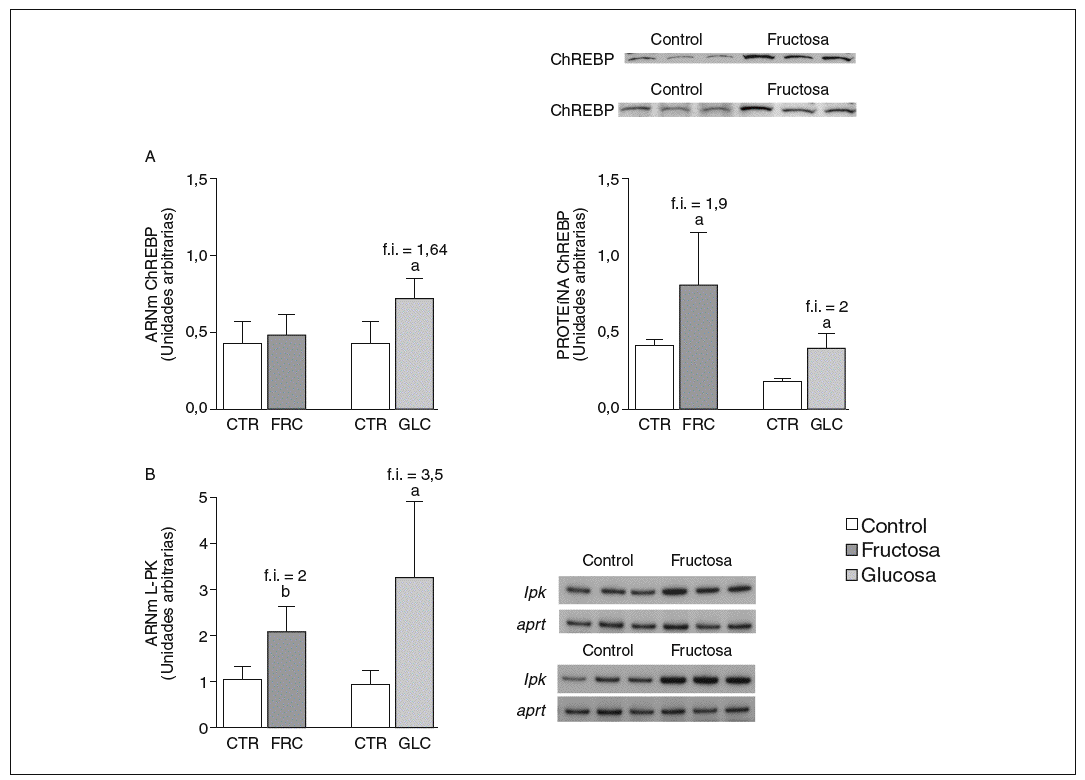
Figura 2. Niveles de expresión hepática de ChREBP y LPK tras la administración de hidratos de carbono. A: niveles relativos de ARNm y de proteína para ChREBP en muestras hepáticas de animales control (CTR) y animales tratados con fructosa (FRC) y con glucosa (GLC). Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. En la parte superior del gráfico derecho se muestra una autorradiografía representativa de la determinación, por Western blot, de los valores de proteína para 3 animales de cada grupo de tratamiento. B: niveles relativos de ARNm para el gen lpk en muestras hepáticas de animales CTR y animales tratados con FRC y GLC. Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. En la parte derecha del gráfico se muestra una autorradiografía representativa de la determinación, por RT-PCR, para 3 animales de cada grupo de tratamiento. Todos los resultados están expresados en unidades arbitrarias. ap < 0,05. bp < 0,01. f.i.: fold induction.
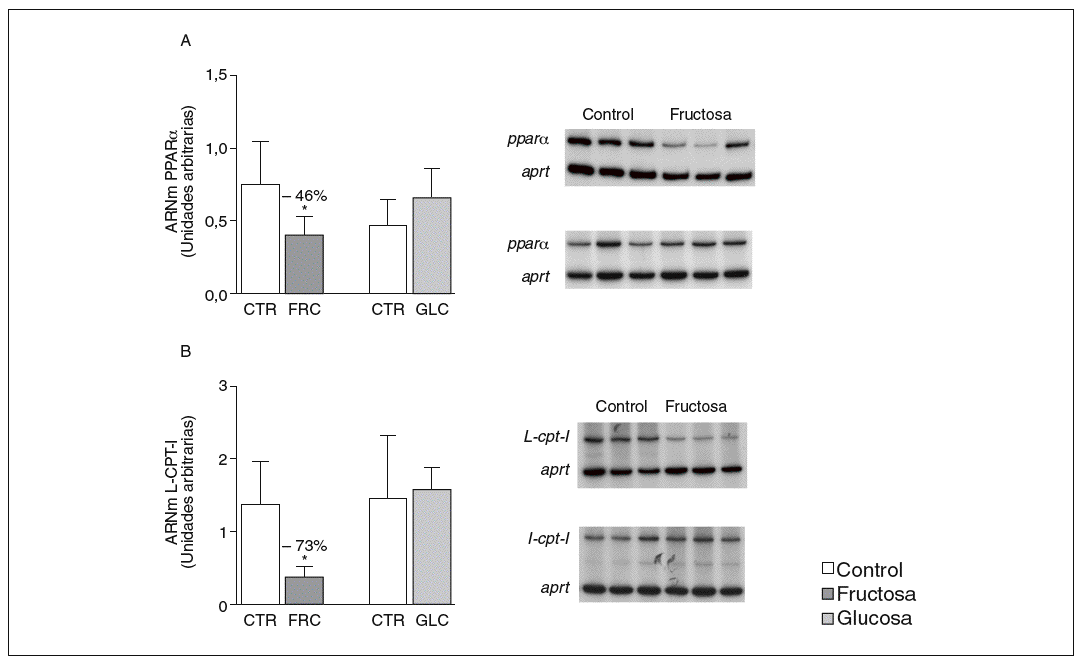
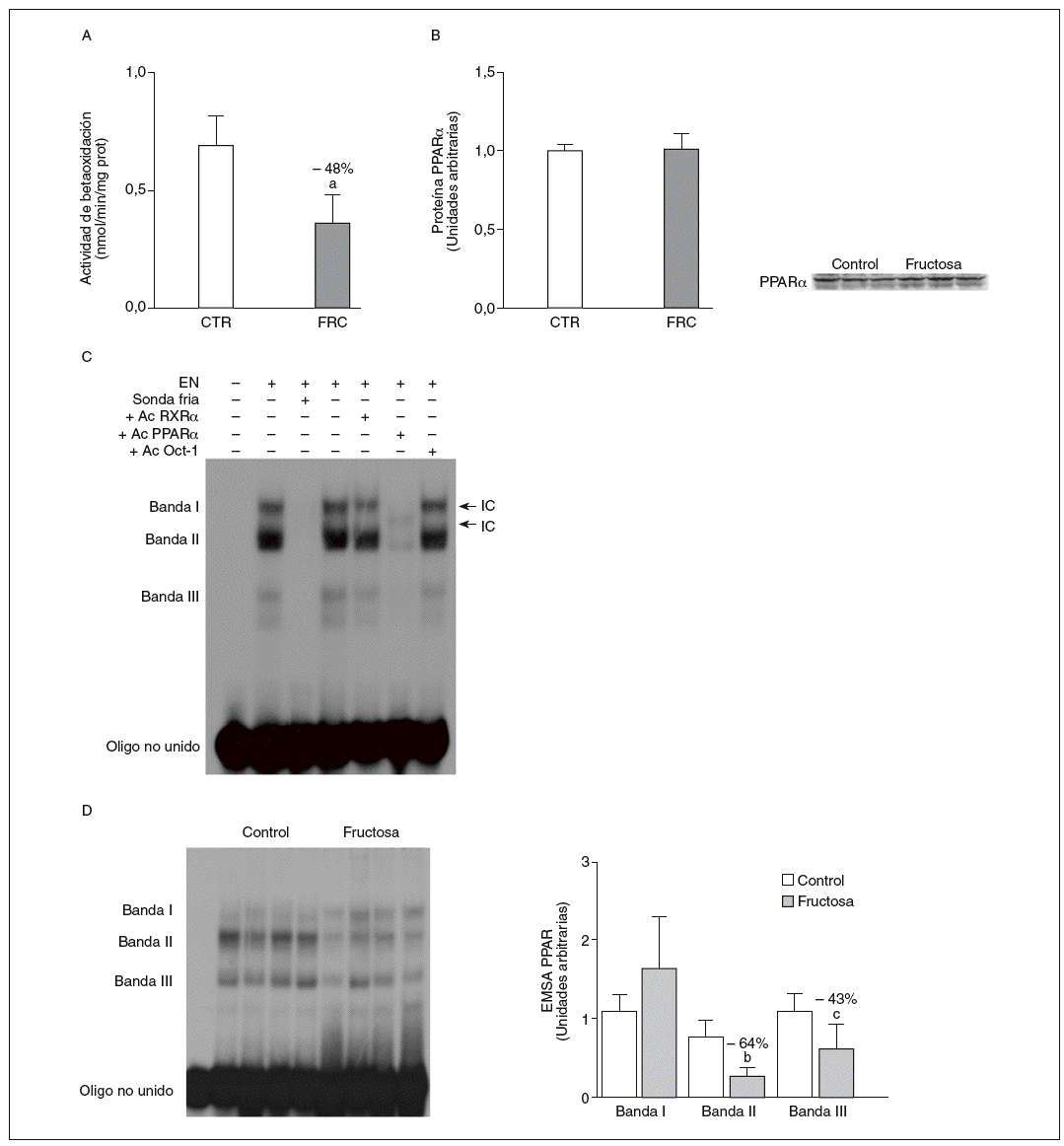
La administración de fructosa produce una marcada reducción de la actividad de betaoxidación hepática de ácidos grasos por disminución de la actividad transcripcional de PPAR*. La administración de fructosa al 10% en el agua de bebida produjo una marcada reducción de los valores de ARNm de PPAR* (46%) y de L-CPT-I (73%). Estos cambios no se observaron en los animales que bebieron glucosa (fig. 3A y B). Como consecuencia de ello, la actividad de betaoxidación hepática de los ácidos grasos disminuyó un 48% en el grupo tratado con fructosa frente al grupo control (fig. 4A). Cuando se determinaron los valores hepáticos de proteína PPAR* no se observaron diferencias entre los animales tratados con fructosa y los controles, lo que sugiere un déficit en la actividad transcripcional de PPAR*, pero no en su expresión hepática (fig. 4B). Por ello se determinó la actividad de unión de extractos nucleares de ratas control y ratas tratadas con fructosa a un oligonucleótido PPRE presente en la región promotora del gen de la L-CPT-I. Tal y como sepecíficas (I, II y III) que en presencia de un exceso de sonda no marcada desaparecen (I, II, III) o se retardan al incubar con un anticuerpo anti-PPAR* (banda II, III); concretamente, la intensidad de la banda II se redujo un 64% y la banda III un 43% en los animales tratados con fructosa, confirmando que ésta es capaz de reducir la actividad trascripcional de PPAR* en el hígado (fig. 4C y D).
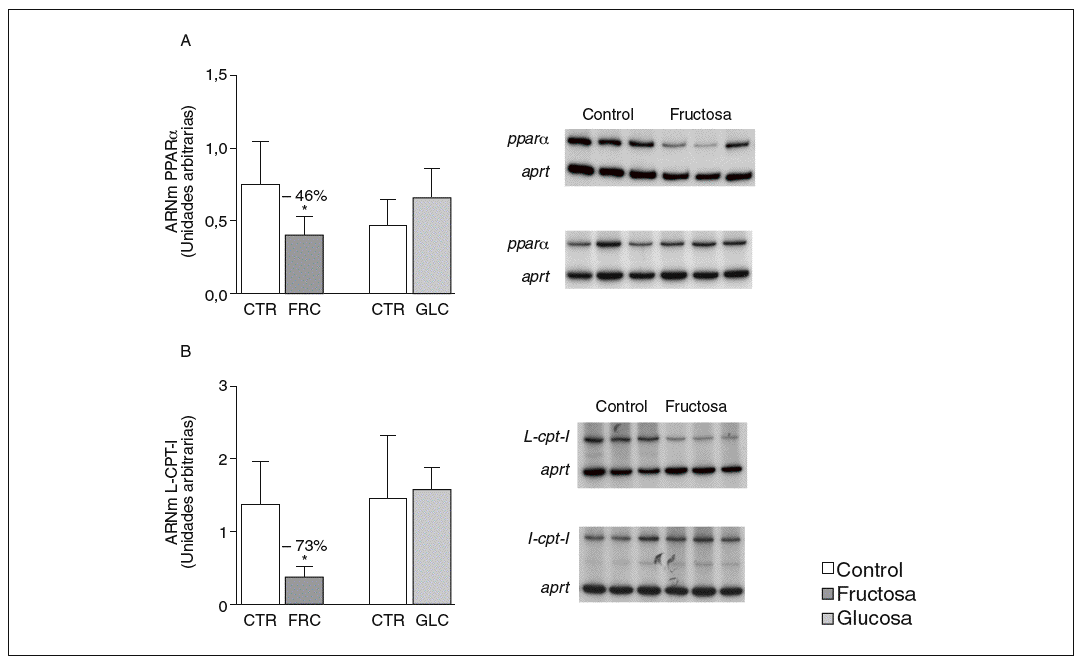
Figura 3. Determinación de los valores de ARNm para PPAR* y L-CPT-I en hígado de ratas alimentadas con hidratos de carbono. A: niveles relativos de ARNm para el gen ppar* en muestras hepáticas de animales control (CTR) y animales tratados con fructosa (FRC) y con glucosa (GLC). Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. En la parte derecha del gráfico se muestra una autorradiografía representativa de la determinación, por RT-PCR, para 3 animales de cada grupo de tratamiento. B: niveles relativos de ARNm para el gen l-cpt-I en muestras hepáticas de animales CTR y animales tratados con FRC y con GLC. Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. En la parte derecha del gráfico se muestra una autorradiografía representativa de la determinación, por RT-PCR, para 3 animales de cada grupo de tratamiento. Todos los resultados están expresados en unidades arbitrarias y se han normalizado por los valores de ARNm del gen control aprt. *p < 0,01.
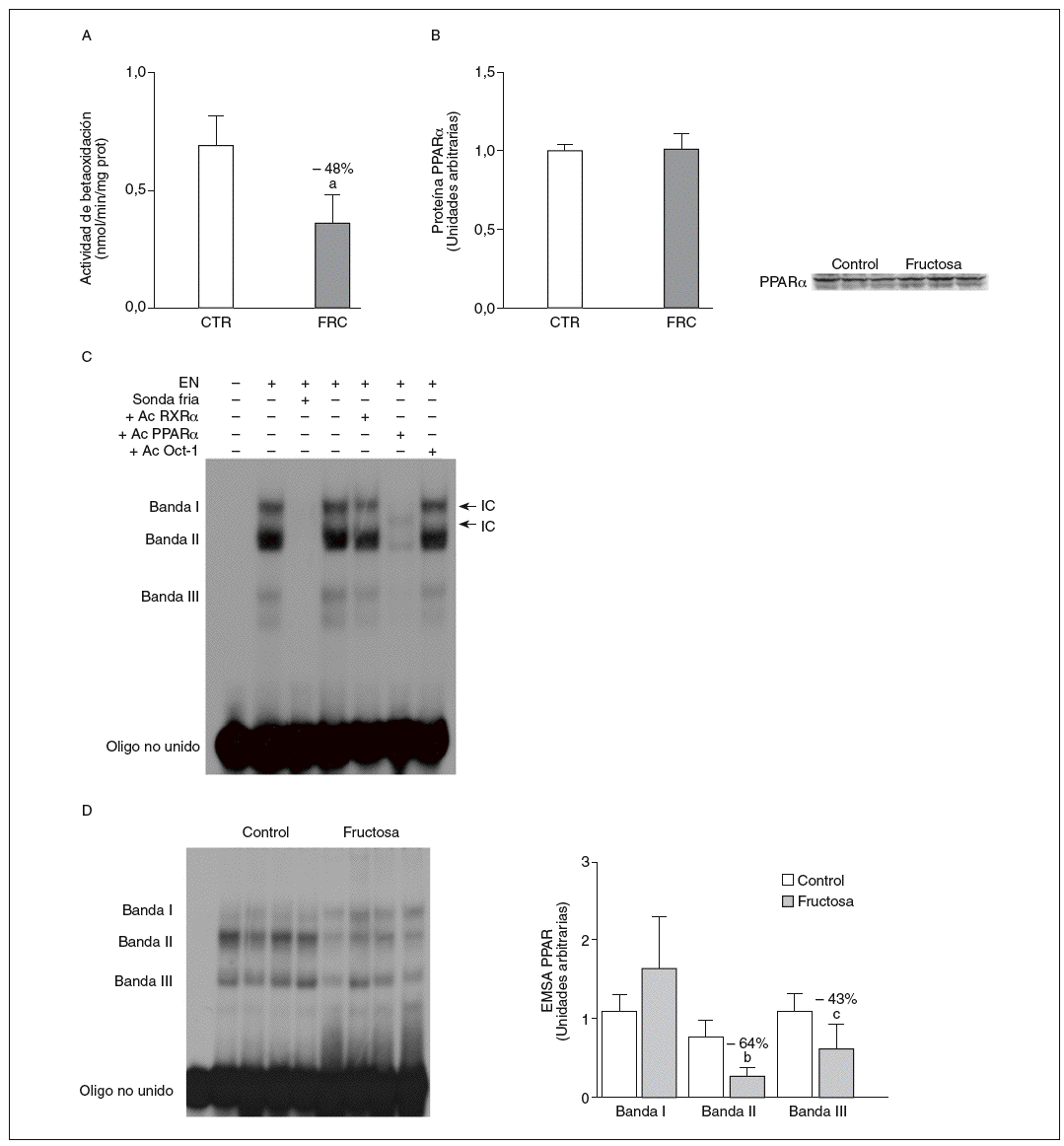
Figura 4. Efecto de la fructosa sobre la oxidación hepática de los ácidos grasos y la actividad de unión PPAR*. A: actividad de betaoxidación de los ácidos grasos expresada en nmol de palmitoil-CoA oxidado/min/mig de sobrenadante posnuclear, en hígado de ratas control (CTR) y ratas tratadas con fructosa (FRC). Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. B: niveles relativos de proteína PPAR* en muestras hepáticas de animales y tratados con FRC. Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. A la derecha del gráfico se muestra una autorradiografía representativa de la determinación, por Western blot, de los valores de proteína para 3 animales de cada grupo de tratamiento. C: ensayo de EMSA donde se muestra la capacidad de unión de los extractos nucleares hepáticos de un animal control al oligonucleótido PPRE, formando 3 bandas específicas de unión (I, II y III). El anticuerpo anti-Oct-I se usó para demostrar que los cambios observados tras la incubación con sonda fría, anticuerpo anti-PPAR* y anti-RXR no eran interferencias inespecíficas. D: autorradiografía representativa del ensayo de EMSA para 4 animales CTR y 4 tratados con FRC donde se muestran las 3 bandas específicas de unión PPRE-extracto nuclear. A la derecha, gráfico con la cuantificación, en unidades arbitrarias, de la intensidad de cada una de las bandas. Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. ap < 0,001. bp < 0,01. cp < 0,05.
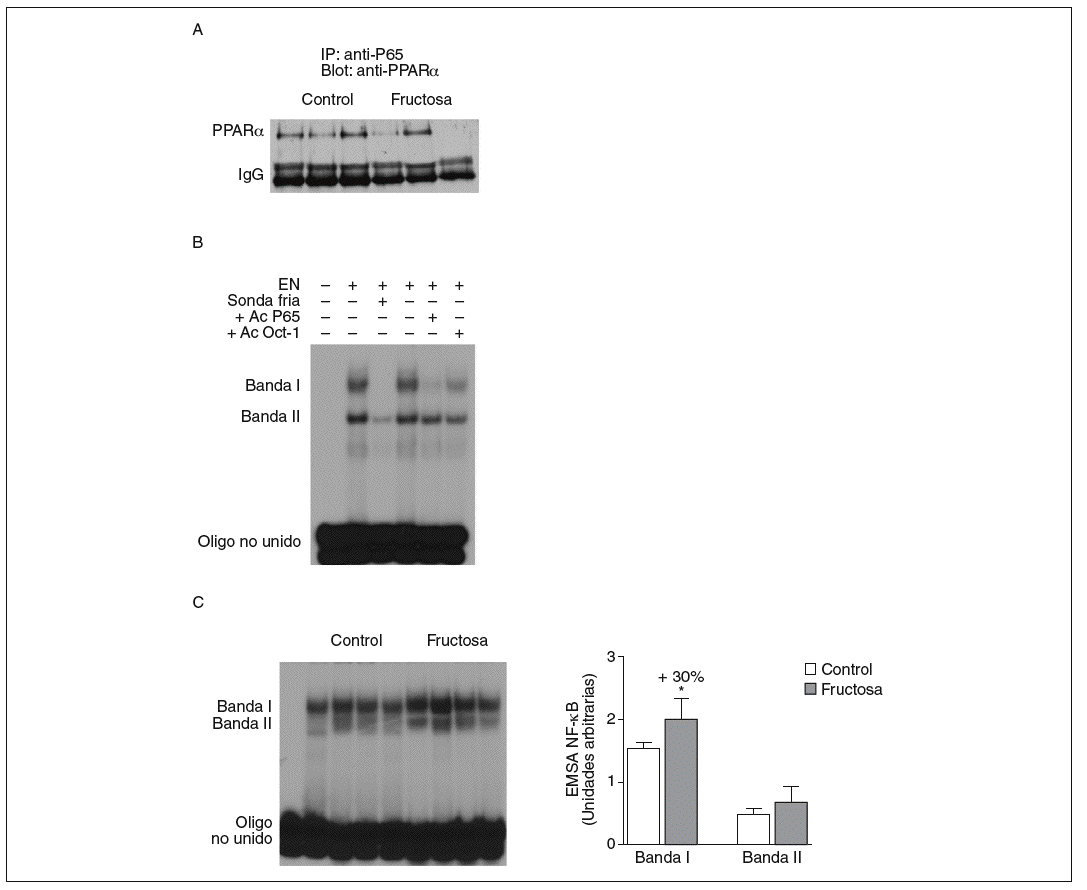
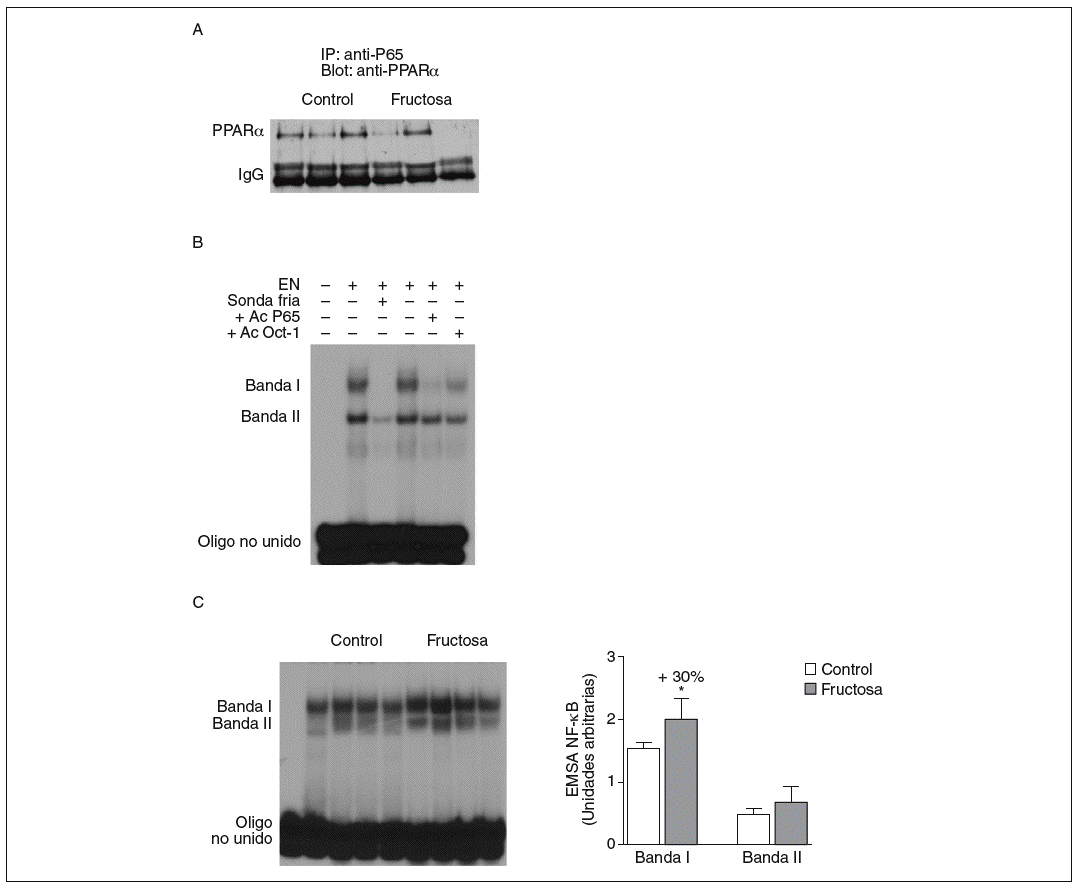
Figura 5. Efecto de la fructosa sobre la actividad transrepresora de PPAR* y NF-*B. A: immunoprecipitación de un anticuerpo anti-p65 con pellets de proteína-A-agarosa y posterior Western blot que revela un anticuerpo anti-PPAR*. Se muestra una autorradiografía representativa del Western blot para 3 animales por grupo de tratamiento. B: ensayo de EMSA donde se muestra la capacidad de unión de los extractos nucleares hepáticos de un animal control al oligonucleótido NF-*B, formando dos bandas específicas de unión (I y II). El anticuerpo anti-Oct-I se usó para demostrar que los cambios observados tras la incubación con sonda fría y anticuerpo anti-p65 no eran interferencias inespecíficas. C: autorradiografía representativa del ensayo de EMSA para 4 animales control y 4 tratados con fructosa donde se muestran las 2 bandas específicas de unión NF-*B-extracto nuclear. A la derecha, gráfico con la cuantificación, en unidades arbitrarias, de la intensidad de cada una de las bandas. Los resultados corresponden a la media ± desviación estándar para n = 6 animales por grupo de tratamiento. *p < 0,05.
La fructosa produce una reducción de la actividad transrepresora de PPAR* en el hígado. El PPAR* también puede unirse físicamente a diferentes factores de trascripción y reprimir su actividad transcripcional. El mecanismo de transrepresión implica una acción bidireccional entre PPAR* y otros factores de transcripción22. Por ello, en nuestro estudio quisimos determinar si la administración de fructosa disminuía la actividad transrepresora de PPAR*, y valorar la interacción fisiológica entre PPAR* y el factor de transcripción NF-*B en extracto nuclear de tejido hepático; el NF-*B es una conocida diana de la actividad transrepresora de PPAR*22. Se realizaron estudios de coinmunoprecipitación (fig. 5A) que mostraron una unión de PPAR* a p65 (uno de los componentes principales de NF-*B) menor en las ratas tratadas con fructosa que en los animales control, lo que indica que la fructosa reduce la actividad transrepresora de PPAR*. Al analizar la capacidad de unión de los extractos nucleares a un elemento de respuesta a NF-*B, aparecieron 2 bandas específicas (I y II) que, en presencia de un exceso de sonda no marcada, desaparecen. Asimismo, al incubar con un anticuerpo anti-p65 la banda I desaparece parcialmente (fig. 5B); concretamente, la intensidad de esta banda I se incrementó un 30% en los extractos nucleares de los animales tratados con fructosa frente a los control, lo que sugiere que la activación de NF-*B por parte de la fructosa se debe a una reducción de la actividad transrepresora de PPAR* en el hígado. Estos resultados no se observaron en las ratas tratadas con glucosa (resultados no mostrados).
Discusión
Estudios previos realizados por nuestro grupo de investigación mostraron que la administración de fructosa al 10% en el agua de bebida producía, en la rata, una reducción de los valores de ARNm de PPAR* y L-CPT-I hepáticos y de la actividad de betaoxidación de los ácidos grasos9. El presente estudio nos ha permitido confirmar los resultados previos y profundizar sobre los posibles mecanismos mediante los cuales la fructosa modifica la expresión hepática de PPAR*.
Hemos demostrado que la administración de fructosa reduce la actividad de transactivación y transrepresión de PPAR* sin modificar su contenido proteico hepático. Como resultado de esta reducción en la actividad transcripcional de PPAR*, los valores de ARNm de sus genes diana, L-CPT-I23 y el mismo PPAR*24, se redujeron. En el caso de L-CPT-I, la reducción del ARNm posiblemente está relacionada con la reducción de los valores de enzima activa y de la oxidación mitocondrial de los ácidos grasos, proceso catabólico directamente controlado por L-CPT-I23. La acumulación de ácidos grasos no metabolizados en el hígado (ligandos naturales de PPAR*)25 es probablemente causante de los valores constantes de proteína PPAR* que se observan en los hígados de las ratas tratadas con fructosa, a pesar de la reducción de los valores de ARNm de este gen, dado que el receptor PPAR* se estabiliza mediante unión al ligando26.
La glucosa, a altas concentraciones, regula negativamente la expresión de PPAR* en células pancreáticas-ß aisladas27. Es posible que los diferentes efectos producidos por los 2 hidratos de carbono sobre el PPAR* en el hígado sean un fenómeno farmacocinético debido a la diferente distribución corporal y de utilización que tienen la fructosa y la glucosa. Mientras la glucosa es usada en distintos tejidos extrahepáticos como fuente de energía, incluidos el cerebro y el músculo esquelético, la fructosa es utilizada casi exclusivamente en el hígado5, hecho que conlleva un mayor metabolismo hepático de la fructosa frente al de la glucosa en este tejido. En concordancia, sólo los animales alimentados con glucosa mostraron un incremento de los valores de AGL plasmáticos, probablemente debido a un cambio en el consumo de ácidos grasos por glucosa como fuente de energía en músculo esquelético, principal tejido consumidor de ácidos grasos.
Nuestros resultados muestran que la administración de fructosa y glucosa en el agua de bebida produce incrementos similares en la expresión de enzimas implicadas en la síntesis de ácidos grasos y de ChREBP. Esto está en concordancia con otros estudios previos que muestran un incremento en la síntesis de novo de triglicéridos VLDL en ratas alimentadas con fructosa o glucosa en el agua de bebida8. Así, el hecho de que sólo los animales que consumieron fructosa presenten hipertrigliceridemia y esteatosis hepática podría atribuirse a la disminución de la oxidación hepática de los ácidos grasos debida al déficit de actividad PPAR*. En situaciones experimentales similares, la reducción de la expresión de PPAR*28-30 o de su actividad transcripcional31 está estrechamente asociada a un incremento de los triglicéridos hepáticos y a hipertrigliceridemia.
La hiperglucemia sostenida promueve el estrés oxidativo y la activación de factores de transcripción proinflamatorios como el NF-*B en células mononucleares32. El consumo de altas cantidades de fructosa (60% p/v en la dieta) también induce al estrés hepático y de otros tejidos junto con la resistencia a la insulina y la desregulación del metabolismo lipídico33. Nuestros resultados indican que la ingesta de fructosa, en ausencia de resistencia a la insulina, es también capaz de producir estrés hepático mediante el incremento de la actividad NF-*B. Esta respuesta es consecuencia de la disminución en la actividad transrepresora de PPAR* producida por la administración del hidrato de carbono.
En las últimas décadas se viene observando una tendencia por parte de la población a aumentar el consumo energético, en forma de dietas hipercalóricas, ricas en fructosa. Este incremento coincide con una mayor prevalencia de obesidad y síndrome metabólico3. Además, la fructosa administrada en forma líquida produce una hipertrigliceridemia mayor que cuando se administra en forma de dieta sólida10. Un incremento del 10% en la ingesta de energía, en forma de hidratos de carbono, no parece ser suficiente para inducir hipertrigliceridemia a no ser que se administre en forma líquida rica en monosacáridos10,34. En una revisión reciente de Bray et al35 se muestra que en modelos de roedor con obesidad y resistencia a la insulina, el consumo de fructosa en el agua de bebida induce al deseo de seguir consumiendo bebidas edulcoradas, reduciendo de forma insuficiente la ingesta de comida sólida, produciendo así un balance calórico positivo y el desarrollo de obesidad. En el presente trabajo, tras 14 días de tratamiento, las ratas que consumieron fructosa no mostraron modificaciones en el incremento de su peso corporal con respecto a las ratas control, posiblemente por la corta duración del experimento; sin embargo, las ratas a las que se administró fructosa comieron menos dieta sólida (13%) que las tratadas con glucosa (22%), respecto los animales control, aun bebiendo la misma cantidad de hidratos de carbono. Nuestros resultados indican que esta situación podría explicarse por un estado de resistencia a la leptina producido por la administración de fructosa, ya que los valores plasmáticos de esta hormona se vieron fuertemente incrementados en el grupo fructosa con respecto el grupo control. Esto nos lleva a pensar que estos cambios observados tras la administración de fructosa en rata serían previos a la aparición de obesidad y resistencia a la insulina, lo que sugiere que el hígado es un órgano clave en el desarrollo de las alteraciones metabólicas producidas por el consumo de fructosa.