










Introducción
La hiperlipidemia familiar combinada (HFC) fue identificada hace más de 25 años como la dislipidemia hereditaria más frecuente entre las familias de pacientes que habían sufrido un infarto de miocardio prematuro. Lo que inicialmente se consideró una alteración que cursaba básicamente con concentraciones elevadas de colesterol y triglicéridos se contempla hoy como un síndrome que también incluye la resistencia a la insulina, la intolerancia a la glucosa, la hipertensión arterial o la obesidad. Es decir, hoy día sabemos que esta enfermedad, caracterizada exclusivamente por los criterios lipídicos que hace un cuarto de siglo sirvieron para definirla, aparentemente afecta a un conjunto heterogéneo de pacientes con distintos fenotipos bioquímicos y metabólicos1.
En esta revisión se discutirán las evidencias clínicas y metabólicas que ponen de manifiesto esta heterogeneidad, los avances en el conocimiento de las bases metabólicas y genéticas de esta enfermedad, su grado de aplicación a la mejora del diagnóstico y las propuestas más recientes para su redefinición.
Fenotipo lipídico
Visión clásica
Identificación de una nueva hiperlipidemia. La existencia de un nuevo fenotipo hiperlipémico (hiperlipidemia combinada) distinto de la hipercolesterolemia o hipertrigliceridemia aisladas fue descrita en 1972 por Rose et al2. Ese mismo año 2 trabajos identificaban este fenotipo como uno de los más frecuentes entre supervivientes de infarto de miocardio prematuro, describían sus características lipídicas y lipoproteicas3,4 y confirmaban lo que estudios realizados 10 años antes ya apuntaban5,6, la hiperlipidemia combinada era el fenotipo presente en un 30% de los supervivientes de un infarto agudo de miocardio3. En el momento de su identificación los análisis lipídicos de control se limitaban a la determinación de colesterol y triglicéridos plasmáticos y se desconocían completamente las alteraciones metabólicas subyacentes a este trastorno. A pesar de la extraordinaria importancia de esta observación, las limitaciones mencionadas obligaron a una definición puramente descriptiva basada en un número limitado de parámetros lipídicos (tabla 1). Aunque el tiempo y los avances científicos han matizado alguna de las observaciones iniciales (alteración monogénica, expresión exclusivamente después de los 20 años), es justo destacar que otras se han visto plenamente confirmadas (defecto básico del metabolismo de los triglicéridos).

Dos eran las características que definían a esta dislipidemia: la existencia de múltiples fenotipos lipoproteicos en las familias de los pacientes y de enfermedad cardiovascular prematura. Más tarde se describió que la variabilidad fenotípica se observaba también en un mismo individuo a lo largo del tiempo7. De hecho, el análisis sistemático de esta variabilidad ha sido clave para una mejor comprensión de los parámetros más fiables para identificar esta alteración.
De esta manera, la alteración denominada hiperlipidemia combinada se diagnosticaba cuando se daban las siguientes circunstancias: a) paciente con fenotipo IIb (hiperlipidemia mixta); b) diferentes fenotipos entre los familiares de primer grado; c) historia familiar de enfermedad arteriosclerótica prematura, y d) exclusión de hiperlipidemias secundarias.
Un hecho importante, que es motivo de reflexión en la actualidad, es que, en los 30 años que han transcurrido desde la identificación de la hiperlipidemia combinada, el criterio diagnóstico ha permanecido prácticamente invariado con una única e importante excepción: la inclusión más o menos generalizada de las concentraciones de apolipoproteína (Apo) B-100. Ello se derivó de 2 observaciones publicadas en 1980: por un lado, la identificación del trastorno denominado hiperapobetalipoproteinemia (concentraciones elevadas de Apo B con colesterol normal)8 y, por otro, la constatación de que los pacientes con HFC presentaban una sobreproducción hepática de Apo B que resultaba en la secreción de un mayor número de lipoproteínas de muy baja densidad (VLDL) y de triglicéridos y que podían cursar con concentraciones normales de colesterol9,10.
En la actualidad, la inmensa mayoría de los estudios relacionados con la HFC utilizan el siguiente criterio en cuanto a parámetros específicos:
Siempre: paciente con hiperlipidemia combinada. Al menos 2 familiares de primer grado con un fenotipo distinto.
No siempre: concentraciones elevadas de Apo B-100. Enfermedad cardiovascular prematura en la familia.
Diagnóstico diferencial. Desde el punto de vista clínico, la HFC se manifiesta con hipercolesterolemia y/o hipertrigliceridemia con fenotipo variable. Como hemos comentado, la coexistencia en familiares de primer grado del paciente índice de dislipidemias similares permite establecer el diagnóstico. Sin embargo, en ocasiones éste no es sencillo debido a que la información sobre la familia no es siempre accesible y a la carencia de un marcador bioquímico definitivo de la entidad. El diagnóstico diferencial se establece en primer lugar con hipertrigliceridemias secundarias a transgresiones dietéticas, consumo de alcohol, obesidad, diabetes, etc., que suelen corregirse al controlar el mecanismo de producción. Sin embargo, y especialmente en el caso de la diabetes, no debe olvidarse que la elevada frecuencia de ambas entidades puede hacer que éstas coincidan y por tanto deberá tenerse presente en el caso de hiperlipidemias difíciles de controlar en pacientes diabéticos. La HFC y la diabetes mellitus 2 se consideran entidades diferenciadas11 y en la actualidad desconocemos si la primera predispone a la segunda más allá de la predisposición que confiere la resistencia a la insulina12.
También debe descartarse el patrón de hipertrigliceridemia con concentraciones bajas de colesterol en las lipoproteínas de alta densidad en pacientes con síndrome metabólico, aunque por lo general cursa con concentraciones de Apo B más bajas. De hecho, la HFC es un trastorno metabólico que forma parte de la constelación del síndrome metabólico, pero su diferente asociación familiar hace que se defina como una entidad distinta aunque comparta en gran medida aspectos patogénicos como la resistencia a la insulina13. La disbetalipoproteinemia o hiperlipidemia tipo III, que puede aparecer en sujetos homocigotos para el genotipo Apo E E2/E2, es la dislipoproteinemia primaria con la que debe establecerse el diagnóstico diferencial. En esta última no suelen encontrarse antecedentes familiares dado su carácter recesivo, pero ya hemos comentado que en ocasiones estos antecedentes son difíciles de averiguar. La determinación del genotipo de Apo E establecerá el diagnóstico.
Visión actualizada
Variabilidad fenotípica y sus implicaciones diagnósticas. Un seguimiento de 20 años de las familias en las que se identificó por primera vez esta alteración14 mostró que un tercio de los familiares cambiaron de fenotipo y que un 17% de los pacientes eran normolipémicos tras el seguimiento. El parámetro menos afectado por la variabilidad lipídica era la Apo B. Los valores de Apo B más altos se detectaban en los individuos que eran persistentemente hiperlipémicos, los valores medios se encontraban en aquellos con fenotipo cambiante y los más bajos en los sujetos exclusivamente normolipémicos. Las lipoproteínas de baja densidad (LDL) pequeñas y densas eran más propias de la hipertrigliceridemia que del fenotipo HFC.
Veerkamp et al15 siguieron durante 5 años a 32 familias diagnosticadas de HFC. Al inicio del seguimiento se consideraron afectados aquellos sujetos con concentraciones de colesterol o triglicéridos superiores al percentil 90 para cada edad y sexo. Utilizando este punto de corte, se ha observado que un 45% de los descendientes de pacientes con HFC son hiperlipémicos16. Tras 5 años, un 25% de los individuos considerados afectados al inicio eran normolipémicos y los parámetros que mejor los diferenciaban de los familiares no afectados eran la Apo B y las LDL pequeñas y densas.
Del Rincón Jarero et al17 llegaron a la conclusión de que unas concentraciones de Apo B entre 115 y 120 mg/dl eran las que mejor se correspondían con la proporción observada de un 50% de afectados por generación (el 55 y el 50%, respectivamente), asumiendo una alta penetrancia de la enfermedad. A concentraciones superiores (130 mg/dl) dicho porcentaje disminuía hasta el 34%.
La consistencia de la Apo B como parámetro característico de esta alteración ha podido apreciarse también en diversos estudios de cosegregación entre las concentraciones de Apo B y el fenotipo HFC18,19.
La variabilidad fenotípica, o más concretamente lipídica, se ha analizado con otros enfoques. Por ejemplo, Delawi et al20 han demostrado que los cambios en el fenotipo lipoproteico que pueden producirse en un paciente con HFC a lo largo del tiempo son debidos especialmente a la elevada variabilidad del colesterol y de los triglicéridos en la fracción VLDL. En consonancia con los hallazgos de estudios anteriores, identifican que uno de los parámetros con menor variabilidad es la Apo B. Lo que resulta llamativo de este trabajo es la constatación de que la variabilidad lipídica en estos pacientes no es superior a la que presentan individuos normolipémicos y que, por tanto, dicha variabilidad no parece ser una característica específica de la HFC. Parece lógico, no obstante, que el hecho de que las concentraciones de colesterol y triglicéridos sean moderadamente elevadas en estos sujetos (y, por tanto, relativamente cercanas al límite de la normalidad) facilite los cambios de fenotipo en comparación con aquellos individuos claramente normolipémicos o hiperlipémicos (más alejados del punto de corte).
En esta misma línea, clásicamente se ha considerado la expresión de la hiperlipidemia antes de los 20 años como factor de exclusión para HFC. Siendo cierto que es infrecuente la aparición de hiperlipidemia declarada a edades tempranas, diversos estudios han mostrado que aproximadamente un 40% de los niños y adolescentes de familias con HFC presentan alguna alteración significativa del perfil lipídico21-23.
Metabolismo
El diagnóstico en la práctica clínica y en la investigación. El criterio diagnóstico de la HFC es fundamentalmente descriptivo, es decir, se basa en la identificación de los parámetros que se hallan alterados en estos pacientes pero no incorpora parámetros que se relacionen con los defectos metabólicos que originan dichas alteraciones.
¿Afecta esto a la identificación de los pacientes cuya hiperlipidemia debe tratarse? Dado que lo indicado es proceder a controlar las concentraciones de colesterol y triglicéridos mediante fármacos y medidas hipolipidemiantes, este criterio de identificación debe considerarse suficiente. Sin embargo, un criterio que cumple su función desde el punto de vista clínico es claramente insuficiente cuando pretendemos seleccionar a pacientes para la investigación de las bases genéticas y metabólicas de la enfermedad. La HFC es una alteración heterogénea y esto significa que diversas alteraciones metabólicas y genéticas acaban dando lugar a la hiperlipidemia mixta que finalmente se detecta. O dicho al revés, que el grupo de pacientes aparentemente homogéneo desde el punto de vista lipídico es heterogéneo en lo que respecta a sus causas tanto genéticas como metabólicas. Este hecho tiene repercusiones obvias no sólo en cuanto a las investigaciones que se llevan a cabo con esta enfermedad, sino también en el ámbito clínico y de tratamiento, ya que parece evidente que un tratamiento será más eficaz cuanto más se dirija a las causas específicas que ocasionan el problema.
Por tanto, se está intentando adaptar el criterio diagnóstico de la HFC para que recoja no sólo las consecuencias (hiperlipidemia mixta) sino también las causas (alteraciones metabólicas). A continuación repasaremos cuáles son las alteraciones metabólicas descritas en estos pacientes y en qué medida podrían incorporarse al criterio diagnóstico.
Visión clásica
Sobreproducción hepática de apolipoproteína
B-100 y lipidemia posprandial. Se acepta que la sobreproducción hepática de lipoproteínas ricas en triglicéridos y de VLDL es la característica fisiopatológica principal de estos pacientes8,10,24-27, o al menos de un amplio subgrupo de ellos, ya que de hecho no existe ningún trabajo que haya valorado su prevalencia de una forma sistemática. Dicha sobreproducción puede presentarse también en normolipidemia (hiperapobetalipoproteinemia). No está claro hasta qué punto la HFC y la hiperapobetalipoproteinemia pueden considerarse entidades distintas28, a pesar de que el riesgo cardiovascular que representan sea significativamente superior en la primera29.
En la HFC el elevado número de partículas ricas en triglicéridos circulantes se acompaña de una incapacidad para hidrolizarlas normalmente Esto es especialmente evidente durante el período posprandial30-33 y tiene una influencia muy importante sobre la pared arterial y el riesgo cardiovascular.
Los quilomicrones nacientes son lipoproteínas aterogénicas34 que, al contrario de las LDL, pueden inducir la formación de células espumosas sin necesidad de ninguna modificación35. En el caso de la HFC, la pared arterial de estos pacientes está expuesta a lo largo del día a concentraciones elevadas de estas lipoproteínas aterogénicas20,30,31,36-40. Esto puede provocar una disfunción de la pared arterial y una acumulación subendotelial de remanentes que conducen a la formación de la placa41.
La coincidencia entre la sobreproducción de Apo B como característica metabólica principal y el hecho de que sea el parámetro más constan-
te dentro de la variación fenotípica de estos pacientes apuntan de manera inequívoca a esta Apo co-
mo parámetro específico en el diagnóstico de la HFC.
¿Qué procesos originan la sobreproducción hepática de Apo B en estos pacientes y de qué manera el conocimiento de dichos procesos puede contribuir a mejorar el diagnóstico de la HFC?
Visión actualizada
La sobreproducción de Apo B da lugar a un mayor pool de lipoproteínas circulantes del tipo VLDL, lipoproteínas de densidad intermedia y LDL, lo cual, como ya se ha dicho, se detecta a través de las concentraciones plasmáticas de Apo B-100. La gran cantidad de VLDL y de lipoproteínas de densidad intermedia plasmáticas compite por la lipólisis con las lipoproteínas de origen exógeno, los quilomicrones, lo cual retrasa significativamente en estos pacientes su retirada del plasma (detectable mediante medición de la Apo B-48, específica de las lipoproteínas posprandiales). Por tanto, otra de las características inequívocamente probadas en estos pacientes es una menor capacidad de eliminar los triglicéridos plasmáticos tras una sobrecarga oral grasa30,31. Este retraso posprandial es también observable en condiciones fisiológicas cuando la lipidemia posprandial se valora mediante la medición de los triglicéridos capilares durante el día20. Algunos estudios apuntan consecuencias interesantes de este retraso posprandial. Por ejemplo, el hecho de que las lipoproteínas ricas en triglicéridos permanezcan un mayor tiempo en la sangre favorecería su unión al endotelio y crearía un pool marginal de lipoproteínas más alto que en individuos sin HFC. El tratamiento con estatinas rebaja la cantidad de lipoproteínas ricas en triglicéridos de este pool marginal de lipoproteínas y contribuye de esta manera a reducir el riesgo de arteriosclerosis en los pacientes con HFC, además de mejorar el perfil lipídico en ayunas40.
Aunque en algunos de estos pacientes puede haber una cierta disminución de la capacidad lipolítica42, se considera que el origen de la sobreproducción hepática de Apo B-100 se halla en una reducida capacidad del tejido adiposo (y tal vez del músculo) para captar los ácidos grasos libres (AGL) procedentes de la lipólisis de los triglicéridos, un fenómeno conocido como "captación" (trapping)43,44. Las causas de esta disfunción pueden ser múltiples, pero tienen una consecuencia bien definida: la derivación hacia el hígado de un flujo de ácidos grasos (medible) y la estimulación de la síntesis hepática de triglicéridos en forma de VLDL. Este flujo de AGL al hígado da lugar, in vivo, a un aumento de la formación posprandial de cuerpos cetónicos (medible)36,39 (fig. 1).

Figura 1. La gestión adecuada de los ácidos grasos libres (AGL) depende en gran medida de su captación periférica por parte de los tejidos adiposo y muscular (trapping). Las rutas y genes implicados en este proceso se detallan en la figura 2. Sin embargo, el control global de los AGL depende de otros procesos como la generación de AGL a partir de la lipólisis de lipoproteínas ricas en triglicéridos (TG), la generación de un flujo de AGL que será dirigido hacia el hígado y los procesos de captación y procesamiento de estos AGL. Una alteración significativa de cualquiera de estos pasos puede favorecer lo que hemos denominado "déficit de gestión de AGL".
MTP: microsomal triglyceride transfer protein; HMG-CoA R: 3-hidroxi-3-metilglutaril coenzima A reductasa; SCD: estearoil coenzima A desaturasa; LPL: lipoproteinlipasa; FAT: fatty acid translocase (CD36); FABP: fatty acid binding protein; ASP: proteína que estimula la acilación (C3adesArg); C5L2: receptor ASP; C3: factor C3 del complemento; HSL: lipasa hormonosensible; aP2: adipocyte fatty acid binding protein; VLDL: lipoproteínas de muy baja densidad; acetil-CoA: acetilcoenzima A; acil-CoA: acilcoenzima A; COL: colesterol; PL: fosfolípidos; EC: ésteres de colesterol.
Son varias las rutas que están implicadas en la captación tisular de AGL (fig. 2)45. La mejor establecida es la promovida por la insulina, ya que está bien descrita la existencia de resistencia a la insulina e hiperinsulinemia en estos pacientes31,46,47. No obstante, esta ruta no es la única y el mal funcionamiento de cualquiera de ellas ocasionará una alteración de la captación periférica de AGL y el consiguiente aumento del flujo de AGL unidos a albúmina hacia el hígado. Éste puede captar los AGL independientemente de los efectos de la insulina. Una vez en el hígado, los AGL se reutilizan para la síntesis de triglicéridos, esterificación de colesterol u oxidación a cuerpos cetónicos. Es importante destacar que triglicéridos y ésteres de colesterol son los máximos determinantes de la síntesis y secreción de VLDL, y que uno de los posibles mecanismos involucrados en el aumento en la síntesis de VLDL en la HFC puede ser la canalización intrahepática de estos AGL.

Figura 2. La captación (trapping) defectuosa por parte de los adipocitos se puede producir por desajustes tanto en las vías controladoras de las captación de ácidos grasos libres (AGL), por ejemplo, insulina o proteína estimuladora de la acilación (ASP), y de la síntesis de triglicéridos (TG) como en los mecanismos controladores de la movilización de dichos TG --p. ej., lipasa hormonosensible (HSL) o catecolaminas--. El resultado en cualquier caso será un mayor flujo de AGL hacia el hígado, que estimulará la síntesis de lipoproteínas de muy baja densidad (VLDL).
TNF: factor de necrosis tumoral; TNF-R: receptor del TNF.
El fenómeno de captación deficiente de AGL podría no ser exclusivo de la HFC. Por ejemplo, se ha detectado en pacientes con lipodistrofia secundaria al tratamiento antirretroviral (Castro Cabezas et al, datos no publicados).
Una de las posibles causas de la captación deficiente de los AGL es una interacción defectuosa entre la proteína que estimula la acilación (ASP) y su receptor (fig. 2). La reciente identificación de dicho receptor48 permitirá localizar posibles alteraciones en su estructura en pacientes con diferentes formas del síndrome metabólico.
El hecho de que la ASP sea estructuralmente idéntica a un producto de la degradación del factor C3 del complemento (C3adesArg) ha dado pie a la observación de que en estado posprandial se producen elevaciones de C337,38,49-52, aunque no de ASP53, lo cual se correspondería con un fenómeno de "resistencia a C3", con un remarcable parecido funcional con la resistencia insulínica e igualmente misterioso en cuanto a sus causas. Este fenómeno se ha observado en pacientes con HFC36-39,49, tanto mujeres como varones39, y se normaliza tras tratamiento con estatinas, aunque no en la misma medida que la hipertrigliceridemia posprandial32,49. En conjunto, estas alteraciones en la activación del factor C3 se han relacionado con el aumento del flujo de AGL al hígado característico de la HFC y con su ineficiente aclaramiento de remanentes aterogénicos posprandiales36,39.
Existen diversos estudios que vinculan a la vitamina A con la HFC54-56, y en este sentido es interesante remarcar que la acción de C3/ASP está controlada por el ácido retinoico57.
Uno de los principales determinantes de la captación es la capacidad de movilizar triglicéridos intracelulares en el tejido adiposo mediante la lipasa hormonosensible. Aunque se detectó menor actividad de esta lipasa in vitro en adipocitos de pacientes suecos con HFC58, esto no pudo corroborarse en otras poblaciones59 ni in vivo60.
Respecto a la lipidemia posprandial, además de la generación de AGL, actualmente conocemos otros efectos negativos como la activación de leu cocitos por parte de partículas ricas en triglicéridos y sus remanentes61,62. Esto da lugar al daño endotelial ocasionado por el estrés oxidativo y a la producción de citocinas derivadas de células proinflamatorias62 que pueden contribuir a la generación de la placa aterosclerótica.
También debe mencionarse un reciente trabajo que identifica a la SCD (estearoil coenzima A desaturasa) como uno de los determinantes más importantes de los triglicéridos plasmáticos, especialmente en la HFC63 y en la resistencia insulínica64. Según estos trabajos, una actividad de la SCD elevada desviaría el flujo de AGL llegados al hígado hacia la síntesis de triglicéridos, en vez de hacia la betaoxidación, lo cual induciría la síntesis de VLDL.
Definición de un nuevo fenotipo: la hipertrigliceridemia-hiperapobetalipoproteinemia
Profundizar en la definición, diagnóstico, patogenia, epidemiología, genética y tratamiento de la HFC ha sido un objetivo constante en el estudio de esta alteración. Desde 1973 se han organizado 3 reuniones científicas internacionales para tratar este tema de forma monográfica. En 1986, en Seattle, se definió con claridad la Apo B como una característica clave en la HFC, aunque esto no significó ningún cambio en el criterio diagnóstico65. Doce años más tarde, una segunda reunión celebrada en Helsinki actualizó los avances en los aspectos genéticos y metabólicos de la HFC, aunque no sirvió para actualizar los criterios diagnósticos sobre la base de dichos avances, cosa que sí tuvo lugar durante la tercera reunión, que se celebró en Barcelona en 2001. Esto se concretó en la definición de un nuevo fenotipo, la hipertrigliceridemia-hiperapobetalipoproteinemia (fig. 3)66. ¿De qué manera se llegó a este fenotipo? Había un claro consenso respecto a las características metabólicas de la HFC más convincentemente demostradas: a) sobreproducción de VLDL; b) LDL pequeñas y densas, y c) lipidemia posprandial retrasada.
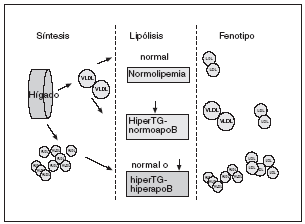
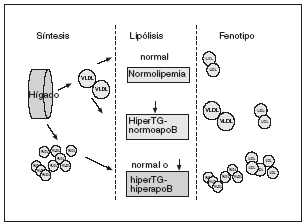
Figura 3. En condiciones normales tanto de síntesis como de lipólisis las lipoproteínas de muy baja densidad (VLDL) se transforman en lipoproteínas de baja densidad (LDL), lo que da lugar a un número de partículas y un contenido de lípidos normales. En una situación de síntesis normal de VLDL y déficit lipolítico se obtendrá un número normal de partículas con un mayor contenido en triglicéridos --hipertrigliceridemia (hiperTG)-normoapobetalipoproteinemia (normoapoB)--. En la hiperlipidemia familiar combinada se parte de una síntesis mayor de VLDL, acompañada o no de un déficit lipolítico, lo cual dará lugar a un número mayor de lipoproteínas (puesto de manifiesto por unas mayores concentraciones de apolipoproteína B) y una mayor cantidad de lípidos, según el grado de deficiencia lipolítica --hiperTG-hiperapobetalipoproteinemia (hiperapoB)--. Se propone que este fenotipo es el que mejor representa el defecto básico de la hiperlipidemia familiar combinada, la sobreproducción hepática de VLDL. Sin embargo, debe quedar muy claro que esta propuesta debe validarse, ya que no está demostrado que efectivamente la hiperTG-hiperapoB sea el fenotipo que mejor identifica a estos pacientes.
La elevada secreción de VLDL-2 da lugar a hipertrigliceridemia y concentraciones elevadas de Apo B. Un mayor tiempo de permanencia de las VLDL-1 resultantes favorece, por un lado, la aparición de LDL pequeñas y densas, y dificulta, por otro, la hidrólisis de lipoproteínas posprandiales. Es decir, la sobreproducción de VLDL explica las características metabólicas de estos pacientes y, por tanto, puede considerarse el defecto primordial de esta alteración. Esto llevó a postular que la hipertrigliceridemia-hiperapobetalipoproteinemia resultante de la sobreproducción de VLDL podría ser un parámetro más representativo y menos cambiante de esta enfermedad.
La propuesta concreta de redefinición del diagnóstico era: a) presencia de hipertrigliceridemia-hiperapobetalipoproteinemia en el paciente y más de un familiar, además de al menos un familiar con enfermedad coronaria prematura, y b) exclusión de hiperlipidemias secundarias, otras alteraciones genéticas y diabetes mellitus 2. Un aspecto destacable de esta propuesta diagnóstica es que la hipercolesterolemia puede estar presente pero no se considera una condición imprescindible para el diagnóstico.
Como ya se ha comentado, esta propuesta se sustenta en avances bioquímicos y metabólicos, pero también analíticos, el más importante de los cuales es la estandarización de la determinación de la Apo B, que actualmente puede medirse con la misma precisión y reproducibilidad que los lípidos plasmáticos67.
Un segundo aspecto clave para el establecimiento de este criterio diagnóstico --y, de hecho, de cualquiera-- es fijar los puntos de corte para las concentraciones de triglicéridos y Apo B. Al principio dichos puntos de corte se fijaron como percentiles poblacionales, con el inconveniente que esto supone dada la variabilidad que existe entre distintas poblaciones. Una alternativa considerada válida fue la de escoger puntos de corte asociados a consecuencias clínicas y fisiopatológicas. En el caso que nos ocupa, se propuso utilizar el criterio de concentraciones de triglicéridos superiores a 1,5 mmol/l, cifra a partir de la cual la presencia de LDL pequeñas y densas aumenta de manera significativa68, y concentraciones de Apo B superiores a 120 mg/dl, que es el valor a partir del cual se aprecia riesgo cardiovascular asociado a hipertrigliceridemia69. Un reciente estudio de seguimiento durante 5 años de familias con HFC concluye que la mejor manera tanto de diagnosticar la enfermedad como de predecir el riesgo cardiovascular que comporta es la basada en las concentraciones de Apo B y triglicéridos junto con el colesterol total ajustado por edad y sexo70.
Genética
Visión clásica
En los primeros estudios y en la mayoría de los trabajos posteriores se observa que aproximadamente el 50% de los familiares de primer grado de estos pacientes tiene concentraciones de colesterol o triglicéridos superiores al percentil 90. Estos datos son consistentes con una transmisión autosómica dominante y, por lo tanto, con la existencia de un gen responsable. A mediados de los noventa una serie de estudios de segregación concluyeron que las concentraciones de colesterol, triglicéridos y Apo B estaban condicionadas por un gen principal, con lo que se reforzaba la idea de un gen único18,71-73. No obstante, este tipo de análisis no permitía concluir si este gen principal era el mismo en cada una de las familias estudiadas.
Se dio así inicio a una larga serie de trabajos que, básicamente mediante 2 estrategias: los estudios de ligamiento y de asociación, han buscado la alteración genética subyacente a la enfermedad y con ella un marcador genético para su diagnóstico. Los primeros intentan localizar un marcador genético que sea capaz de discriminar dentro de una familia a los sujetos afectados de los no afectados. Los estudios de asociación se basan en establecer a escala poblacional (casos y controles) relaciones entre marcadores genéticos de genes candidatos y distintos parámetros relacionados con la enfermedad.
Una vez identificadas la sobreproducción de VLDL, la elevación de las concentraciones de Apo B y las LDL pequeñas y densas como características de la HFC, gran parte de los estudios se dirigieron al análisis del gen de la Apo B, con resultados claramente negativos74,75. De manera global, los resultados obtenidos durante este tiempo se centran en 2 regiones de interés (los cromosomas 1 y 11) y aportan la siguiente información: no existe un gen principal que explique la HFC de forma generalizada.
El primer marcador capaz de discriminar de manera significativa entre individuos afectados y no afectados dentro de una familia con HFC se identificó en la región cromosómica 11q-23-24, donde se codifican las Apo AI-CIII-AIV76. Desafortunadamente, este resultado no pudo reproducirse en otras poblaciones, con lo que se desvaneció la posibilidad de contar con un marcador genético para la HFC77,78. No fue hasta 1998 cuando hubo un avance significativo en este campo, con la identificación de ligamiento positivo entre la región 1q21-23 y la HFC79, que además era un marcador válido en un modelo murino Hyplip de la enfermedad80. Estos resultados se han reproducido en otras poblaciones81,82, lo cual es esperanzador. Por tanto, es razonable suponer que esta región aloja un gen relevante para la etiopatogenia de la HFC.
Recientemente se ha identificado un segundo locus, el Hyplip2, que predispone a la hiperlipidemia combinada y a la arteriosclerosis en ratones83. La identificación del gen homólogo en humanos aportará un nuevo gen candidato para la HFC.
Sí existen genes moduladores de la hiperlipidemia en estos pacientes.La detección de estos marcadores ha permitido identificar una serie de genes moduladores con una influencia significativa en la regulación de las concentraciones lipídicas en estos pacientes. Especialmente relevante es la región del agregado AI-CIII-AIV, concretamente el gen de la Apo C-III. Variaciones de este gen han demostrado ejercer una clara influencia en la inducción de hipertrigliceridemia en pacientes con HFC77,84-87. Este papel modulador se ha visto considerablemente reforzado con la reciente identificación del gen de la Apo A-V88, que ha demostrado ser un potente inductor de hipertrigliceridemia en ayunas en estos pacientes89.
No obstante, ninguno de estos hallazgos ha tenido hasta el momento ninguna repercusión en la mejora del diagnóstico de la HFC, y por tanto puede afirmarse con certeza que no existe ningún marcador genético específico para la HFC en la actualidad90.
Visión actualizada
Respecto a la búsqueda de un marcador genético, los numerosos estudios de ligamiento llevados a cabo han demostrado claramente que no hay una alteración genética única que explique este trastorno. Los marcadores identificados, entre los que cabe destacar las ya mencionadas regiones 11q23-24 y 1q21-23, muestran tasas de asociación con la enfermedad que difieren de forma muy importante entre familias y entre poblaciones. Esta circunstancia se debe en parte a la heterogeneidad genética de esta enfermedad, sin duda acentuada por factores ambientales, pero éste no es el único factor que explica tanta heterogeneidad. Dado que los estudios genéticos, especialmente los de ligamiento, pero también los de asociación, se basan en la determinación inequívoca de individuos afectados y no afectados, la incapacidad del criterio diagnóstico actual para identificar con exactitud a los sujetos afectados, o de identificar a aquéllos con la misma alteración, supone un problema de primera magnitud en este tipo de análisis. En la figura 4 se ejemplifica el impacto que el criterio diagnóstico puede tener en la interpretación de resultados genéticos (Ribalta et al, datos no publicados).

Figura 4. La hipertrigliceridemia-hiperapobetalipoproteinemia (hiperTG-B) y la hiperlipidemia combinada empleada para diagnosticar la hiperlipidemia familiar combinada (HFC) probablemente identifican trastornos metabólicos distintos, al menos en algunos casos. Ello es especialmente relevante en los estudios genéticos, en los que tiene una gran importancia la asignación de los sujetos estudiados a categorías fiables. En esta figura se muestra cómo, al estudiar determinados polimorfismos en el mismo grupo de pacientes con HFC identificados según el criterio diagnóstico habitual o según el criterio de la hiperTG-B, se obtienen discrepancias que en algún caso alcanzan hasta el 21%.
Respecto al papel de los genes moduladores, en general no son específicos de la HFC, sino que son los mismos que predisponen a la hiperlipidemia en la población general91. Sin embargo, cabe hacer 2 matizaciones: a) en algunos casos la frecuencia de estos marcadores está aumentada en pacientes con HFC, lo que indica que tienen una contribución significativa en el desarrollo de la enfermedad, y b) el efecto de estos polimorfismos suele ser mayor en los pacientes con HFC que en los portadores normolipémicos, pero en la misma medida en que el efecto es mayor en sujetos obesos o diabéticos.
Podríamos definir como genes moduladores susceptibles de implementar el diagnóstico de la HFC aquellos que: a) modifican significativamente alguno de los parámetros característicos de la HFC (triglicéridos y Apo B), y b) se presentan con mayor frecuencia entre estos pacientes que en la población general, lo cual indica que su influencia es significativa en el desarrollo de la enfermedad.
Aunque existe un buen número de genes cuyas variaciones modifican el metabolismo lipoproteico (Apo B-100, Apo E, Apo C-III, Apo A-IV, Apo A-V, lipoproteinlipasa, lipasa hepática, proteína transportadora de colesterol esterificado, receptor de las LDL), en la mayoría de los casos la rareza de estas variaciones hace que sean una herramienta poco útil para implementar el diagnóstico de la HFC91. Sobre la base de estas consideraciones, sólo 3 genes presentan variaciones lo suficientemente frecuentes y con un efecto suficientemente significativo para cumplir esta función: la Apo E, la Apo C-III y la Apo A-V (tabla 2)84-86,89,92,93.

Los avances en el conocimiento del metabolismo de la HFC son clave de cara a nuevos enfoques de los estudios genéticos. Las coincidencias entre la HFC y otras expresiones del síndrome metabólico como la diabetes mellitus tipo 2 o la lipodistrofia son incuestionables, y este punto se halla sólidamente respaldado por los estudios genéticos que indican que la región 1q21-23 no sólo presenta ligamiento positivo con la HFC, sino también con la diabetes mellitus tipo 2 y la lipodistrofia94,95. Inicialmente se pensó que el gen de la TXNIP (thioredoxin interacting protein), que es uno de los principales reguladores del estado de oxidorreducción celular y tiene una influencia directa sobre el metabolismo de los AGL, podía explicar esta asociación96. Sin embargo, recientemente se ha identificado el gen que codifica para el factor de transcripción USF-1 (upstream transcriptor factor 1), que regula varios genes relacionados con el metabolismo de los lípidos y la glucosa, como el responsable del ligamiento entre HFC y 1q21-2397.
Por tanto, cabe pensar que la búsqueda de marcadores genéticos podría orientarse a un fenotipo más amplio que podríamos denominar "déficit de gestión de AGL". Tal como ilustra la figura 5, existe un gran número de causas potenciales de este síndrome y, si estudiáramos a un grupo de pacientes con déficit de gestión de AGL, podría ser que cada uno de ellos lo padeciera por un error genético distinto.

Figura 5. La captación (trapping) deficiente, o lo que hemos denominado en esta revisión "déficit de gestión de ácidos grasos libres" (DGA), puede ser el principal defecto de la hiperlipidemia familiar combinada. El fenotipo final será el resultado de un elevado número de factores, entre ellos: a) el defecto específico responsable de los ácidos grasos libres. Lógicamente habrá distintas causas que originarán distintos grados de disfunción; b) la existencia del fondo genético individual hará que esta disfunción se exprese como un defecto más lipídico, tendente a la obesidad, de mal control de la glucosa, y c) los 2 factores mencionados estarán fuertemente modulados por un gran número de factores ambientales que tendrán una gran influencia sobre el fenotipo final.
ASP: proteína que estimula la acilación; RI: resistencia a la insulina; TNF: factor de necrosis tumoral; LPL: lipoproteinlipasa; HL: lipasa hepática; Glut: transportador de glucosa; PPAR-*: receptor activado por proliferadores peroxisómicos-*; UCP: proteínas desacopladoras; HTA: hipertensión arterial; SRA: sistema renina-angiotensina; AGT: angiotensina; ECA: enzima de conversión de la angiotensina; NOs: óxido nítrico sintetasa; PUFA: ácidos grasos poliinsaturados; MUFA: ácidos grasos monoinsaturados; FAT: fatty acid translocase; FATP: transportadores de ácidos grasos; FABP: fatty acid binding protein; aP2: proteína específica de adipocito 2 de los ácidos grasos; TXNIP: thioredoxin interacting protein; HSL: lipasa hormonosensible; RA: ácido retinoico.
Varios de estos genes ya se han explorado en relación con la HFC, por ejemplo, los relativos a la ruta de la insulina, los receptores adrenérgicos o la lipasa hormonosensible, y en todos ellos el resultado ha sido negativo98. Otros, como el receptor C5L2 de la ASP, son objeto de investigación en la actualidad.
Hasta hace poco esta multiplicidad de genes candidatos suponía una clara limitación para la investigación genética. Sin embargo, las nuevas tecnologías que, como los microarrays, permiten examinar a la vez un número elevado de genes nos aportan una potente herramienta con la que abordar estos nuevos enfoques.
El futuro del diagnóstico de la hiperlipidemia familiar combinada
El futuro del diagnóstico de la HFC pasa por intentar identificar las causas y definir con mayor precisión las consecuencias. Existe base científica suficiente para postular que el origen de la hiperlipidemia de estos pacientes está en la sobreproducción hepática de VLDL, y los datos de que disponemos inducen a pensar que ésta está causada por una gestión deficiente de los ácidos grasos en los tejidos periféricos, junto con una excesiva canalización hepática hacia la síntesis de triglicéridos. No disponemos aún de un buen marcador bioquímico para esta captación deficiente, aunque las concentraciones de AGL y la producción posprandial de cuerpos cetónicos son indicativas de este trastorno. El resultado es un aumento de VLDL que se manifiesta en concentraciones elevadas de triglicéridos y Apo B, la hipertrigliceridemia-hiperapobetalipoproteinemia, que se propone como uno de los fenotipos lipídicos de la HFC. Esto hace pensar que tal vez en el futuro habrá que identificar a los individuos con un déficit de base en el manejo de los AGL, lo cual tendrá una expresión variable según la dotación de genes moduladores, su estilo de vida, etc. (fig. 5).
¿Quiere esto decir que la HFC no existe como entidad diferenciada? Su particular asociación familiar respecto a otras enfermedades o el hecho de que, por ejemplo, las concentraciones de Apo B sean más elevadas en estos pacientes que en sujetos con la misma grasa visceral, índice de masa corporal o sensibilidad a la insulina99 más bien hacen suponer que la HFC es la expresión más lipídica del síndrome de déficit de gestión de los AGL.
¿Cuál es entonces el camino que debe seguir el diagnóstico de la HFC para ser más específico? Sin ninguna duda, la genética y la bioinformática han de desempeñar un papel clave en este aspecto, en primer lugar, ayudando a diagnosticar el déficit de gestión de AGL mediante análisis por arrays de todos los genes que puedan alterar el manejo de los AGL, y en segundo lugar, analizando la predisposición individual a cada una de las expresiones de este síndrome que hacen que se vean alterados de manera más importante el metabolismo de los lípidos, el de la glucosa o la diferenciación del tejido adiposo. Dicha predisposición deberá tener en cuenta una amplísima batería de genes moduladores y una no menos extensa lista de factores ambientales.
Las repercusiones de esta mejora diagnóstica no son sólo teóricas y con toda seguridad tendrán un fuerte impacto en el campo terapéutico. Este enfoque diagnóstico lleva implícita la identificación de las causas del fenotipo resultante, lo cual sienta obviamente las bases para un tratamiento más efectivo.
Por último, desde el punto de vista de la prevención cardiovascular hay que subrayar que el fenotipo de hipertrigliceridemia-hiperapobetalipoproteinemia identifica en cualquier caso a sujetos con un riesgo elevado de arteriosclerosis100. Más allá de los matices genéticos y metabólicos que permitan una posterior clasificación en HFC o síndrome metabólico, debemos constatar que se trata de un fenotipo muy frecuente que debe tratarse más enérgicamente si de verdad queremos rebajar el riesgo general de arteriosclerosis.