




Osteoporosis is a systemic skeletal disease, characterized by low bone mass and deterioration in the micro-architecture of bone tissue, which causes increased bone fragility and consequently greater susceptibility to fractures. It is the most frequent metabolic bone disease in our population, and fractures resulting from osteoporosis are becoming more common. On the other hand, vascular calcification is a recognized risk factor of cardiovascular morbidity and mortality, that historically has been considered passive and degenerative process. However, it is currently recognized as an active process, which has histopathological characteristics, mineral composition and, initiation and development mechanisms characteristic of bone formation. Paradoxically, patients with osteoporosis frequently show vascular calcifications. Traditionally, they have been considered as independent processes related to age, although more recent epidemiological studies have shown that there is a close relationship between the loss of bone mass and vascular calcification, regardless of age. In fact, both entities share risk factors and pathophysiological mechanisms. These include the relationship between proteins of bone origin such as osteopontin and osteoprotegerin with vascular pathology, and the protein intercellular system RANK/RANKL/OPG and the Wnt signaling pathway. The mechanisms linked in both pathologies should be considered in clinical decisions, given that treatments for osteoporosis could have unforeseen effects on vascular calcification, and vice versa. In short, a better understanding of the relationship between both entities can contribute to propose strategies to reduce the increasing prevalence of vascular calcification and osteoporosis in the aging population.
La osteoporosis es una enfermedad esquelética sistémica, caracterizada por baja masa ósea y deterioro en la microarquitectura del tejido óseo, que origina un aumento de la fragilidad ósea y en consecuencia mayor susceptibilidad a fracturas. Es la enfermedad metabólica ósea más frecuente en nuestra población y, las fracturas resultantes de la osteoporosis son cada vez más comunes. Por otro lado, la calcificación vascular es un factor de riesgo reconocido de morbilidad y mortalidad cardiovascular, que históricamente era considerada como un proceso pasivo y degenerativo. Sin embargo, en la actualidad se reconoce como un proceso activo, que tiene características histopatológicas, de composición mineral y de mecanismos de iniciación y desarrollo propias de la formación del hueso. Paradójicamente, los pacientes con osteoporosis muestran con frecuencia calcificaciones vasculares. Tradicionalmente se han considerado como procesos independientes relacionados con la edad, aunque estudios epidemiológicos recientes han evidenciado que existe una estrecha relación entre la pérdida de masa ósea y la calcificación vascular, independiente de la edad. De hecho, ambas entidades comparten factores de riesgo y mecanismos fisiopatológicos. Entre ellos destacan la relación entre proteínas de origen óseo como la osteopontina y la osteoprotegerina con la patología vascular, y el sistema intercelular proteico RANK/RANKL/OPG y la vía de señalización Wnt. Los mecanismos vinculados en ambas patologías deben considerarse en las decisiones clínicas, dado que los tratamientos para la osteoporosis podrían tener efectos imprevistos en la calcificación vascular, y a la inversa. En definitiva, una mejor comprensión de la relación entre ambas entidades puede contribuir a plantear estrategias para disminuir la prevalencia creciente de calcificación vascular y osteoporosis en la población que envejece.
Osteoporosis is a systemic skeletal disease, characterised by low bone mass and deterioration in bone tissue microarchitecture, which causes increased bone fragility, with the consequent increase in the risk of fracture.1 It is a serious public health problem, since it affects more than 200 million people. The annual incidence of hip fracture is 1.7 million worldwide. This pathology in turn implies an increase in morbidity and mortality in these patients, so that women with a hip fracture have a mortality of 10–20% higher than expected for their age. By 2050, it is estimated that the annual incidence of hip fracture will increase, with a total of 6.3 million affected, mainly due to the ageing of the world population and the increase in the incidence of falls in these individuals.2 Bone is a connective tissue with high metabolic activity that is under a constant process of renewal. Osteoporosis is caused by an imbalance between bone formation and reabsorption, the responsibility of osteoblasts and osteoclasts, respectively.3 Paradoxically, patients with osteoporosis frequently show vascular calcifications.4
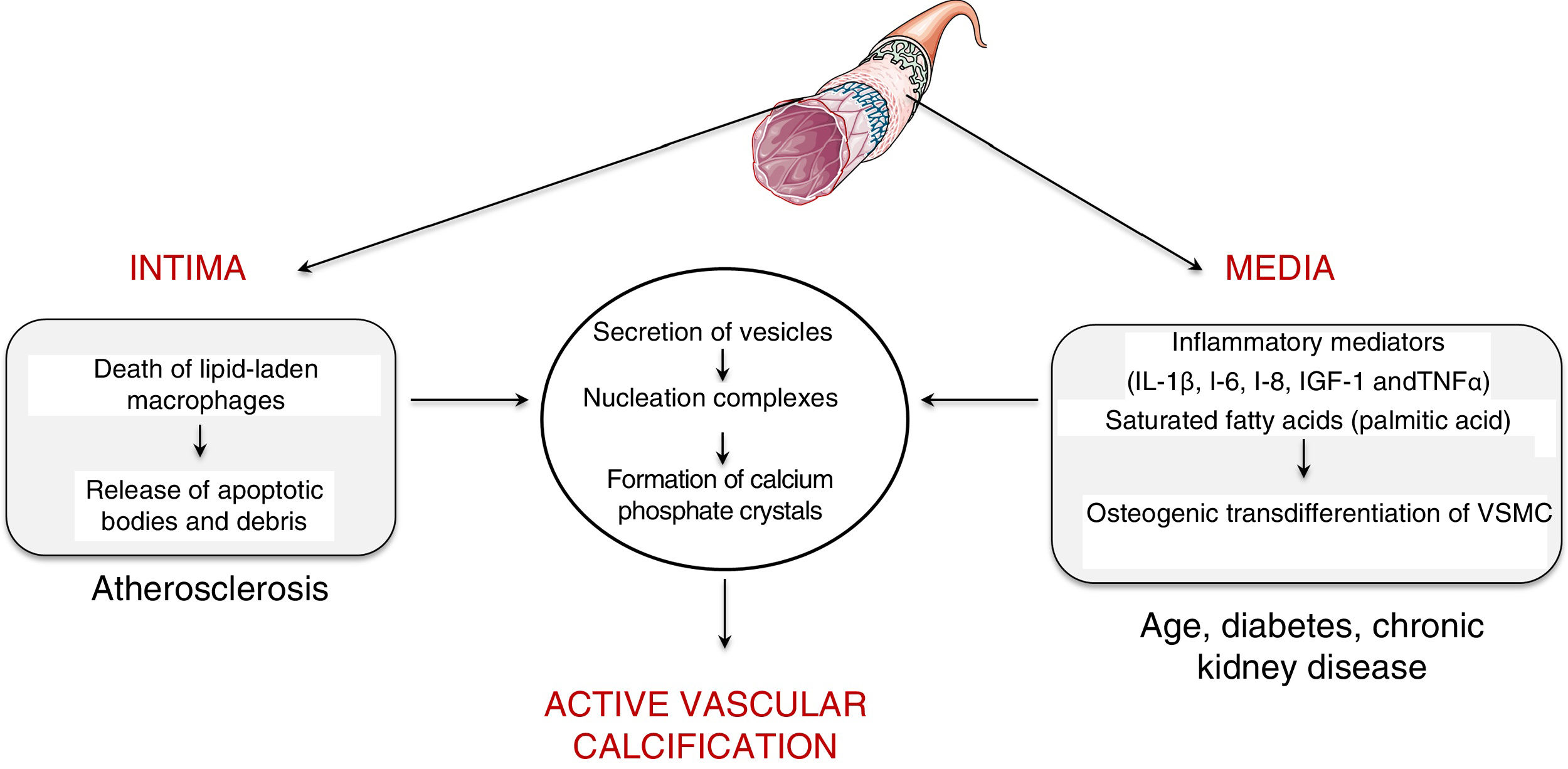
Vascular calcificationTypes and mechanismsVascular calcification was first described by the pathologist Rudolph Virchow in 1863, who described it as a process similar to ossification.5 In fact, vascular calcification shares a series of histopathological characteristics, mineral composition and mechanisms of initiation and development that characterise bone formation. Two processes capable of inducing vascular calcification have been described, one passive process and another active process. Passive calcification is a process independent of cellular activity and results from the deposition of calcium and phosphate ions on elastin and the collagen fibrils that make up the vessel due to the affinity of charge.6 Specifically, calcium ions have spontaneous affinity for elastin and collagen binding sites. These structures, which initially have a neutral charge, are positively charged and attract phosphate and carbonate ions with a negative charge, which initiates the mineralisation process. This phenomenon leads to the formation of hydroxyapatite crystals and the consequent calcification of the extracellular matrix in an amorphous manner. On the contrary, active calcification depends on cellular activity, where chronic inflammation plays a key role.7 In fact, the causal relationship between vascular inflammation and calcification has been clinically proven.8 Specifically, it has been observed by positron emission tomography that vascular inflammation precedes vascular calcification.8 Active calcification can be triggered in both the intimal and medial layers of the vascular wall (Fig. 1). While intimal calcification is associated with the development of atherosclerosis, calcification of the media is associated with age, diabetes and chronic kidney disease.6
 and the media (commonly associated with age, diabetes and chronic kidney failure). VSMC: vascular smooth muscle cells.")
Intimal calcification is largely determined by the inflammatory response that governs the development of atherosclerotic disease. Briefly, the development of the atherogenic process is initiated by the retention of low-density lipoproteins (LDL) in the subendothelial extracellular matrix.9 These retained lipoproteins undergo modifications (mostly associated with oxidation) and are capable of activating the vascular endothelium by inducing the expression of adhesion molecules, cytokines and growth factors that initiate the recruitment of proinflammatory cells. Attracted monocytes are internalised in the vascular intima, where they differentiate into macrophages and are able to phagocyte and accumulate the cholesterol of oxidised lipoproteins becoming foam cells.10 When the phagocytic capacity of the foam cells is exceeded by the amount of cholesterol, their cell death is triggered, with the consequent release of apoptotic bodies and necrotic remains, which serve as nucleation centres for the formation of calcium phosphate crystals.
As for the calcification in the medial layer, it is believed that it is largely determined by the presence and activity of inflammatory mediators. Several studies have established that inflammatory cytokines — interleukin (IL)-1β, IL-6, IL-8—, insulin-like growth factor-1 and tumour necrosis factor (TNF)-α are able to induce in vitro osteogenic differentiation of vascular smooth muscle cells (VSMC). In this way, the presence of an inflammatory environment can trigger an osteogenic transformation of VSMC. In the same way, it has also been described that saturated proinflammatory fatty acids, such as palmitic acid, are capable of directly inducing the transdifferentiation of VSMC towards an osteoblastic phenotype. As with bone formation, both macrophages/foam cells in apoptotic state present in the atherosclerotic plaque and osteogenic vascular cells have the ability to release calcifying vesicles and thus contribute to the initial stages of intimal and medial calcification, respectively. Specifically, it has been suggested that they release vesicles containing lipids (phosphocholine and phosphoethanolamine), proteins (annexins) and ionic components (Ca2+ and inorganic phosphate) necessary to form membrane anchored structures that are the starting point for the formation of calcium phosphate minerals, a process known as nucleation (for more details, see Krohn et al.11).
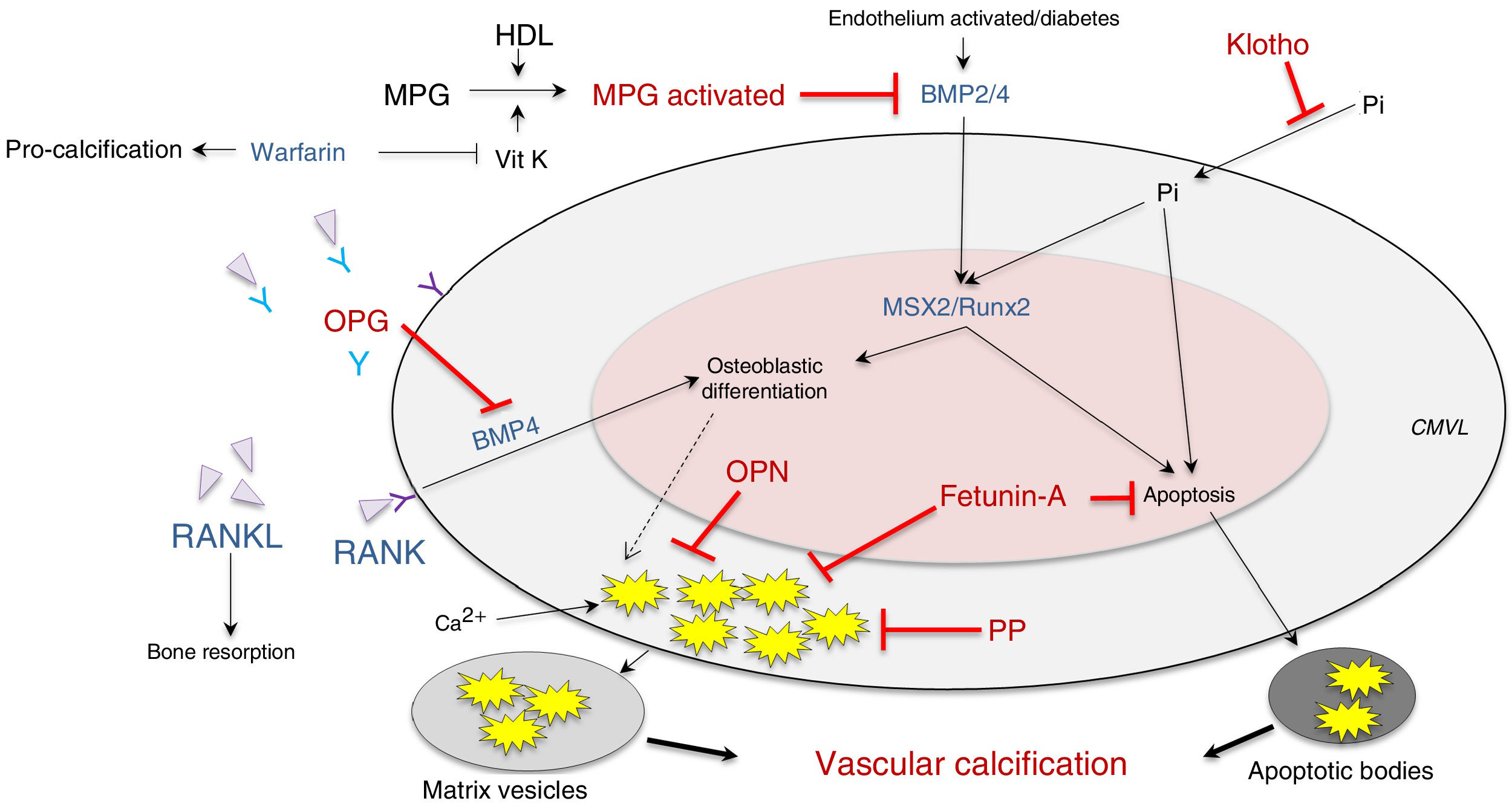
It has also been postulated that vascular calcification occurs when there is increased activity in calcification inducers - receptor activator of nuclear factor κ B/receptor activator of nuclear factor κ B ligand/osteoprotegerin (RANK/RANKL/OPG), bone morphogenetic protein (BMP)2/4 and transcription factors (transcription factor related to runt [RUNX-2] and homeobox protein [MSX2])—, as well as alterations in its inhibitors — acidic protein γ-carboxyglutamic (MGP), pyrophosphate, fetuine-A, osteopontin (OPN), Klotho and osteoprotegerin—, which are expressed constitutively both in the tissue and at the circulating level (Fig. 2; modified from Nakahara et al.12).
 and inducers (blue) of vascular calcification at the level of vascular smooth muscle cells. BMP: bone morphogenetic protein 2/4; HDL: high-density lipoproteins; MGP: matrix Gla (g-carboxyglutamic acid) protein; MPG: receptor activator of nuclear factor-κB ligand (RANKL); MSX2: transcription factor related to the homeobox protein; OPG: osteoprotegerin; OPN: osteopontin; PP: pyrophosphate; RUNX-2: runt-related transcription factor; VitK: vitamin K; VSMC: vascular smooth muscle cells.")
Inhibitors (red) and inducers (blue) of vascular calcification at the level of vascular smooth muscle cells.
BMP: bone morphogenetic protein 2/4; HDL: high-density lipoproteins; MGP: matrix Gla (g-carboxyglutamic acid) protein; MPG: receptor activator of nuclear factor-κB ligand (RANKL); MSX2: transcription factor related to the homeobox protein; OPG: osteoprotegerin; OPN: osteopontin; PP: pyrophosphate; RUNX-2: runt-related transcription factor; VitK: vitamin K; VSMC: vascular smooth muscle cells.
Impact of cardiovascular risk factors on coronary calcification and risk of events
In 1964, a pioneering study published in Lancet13 reported that the calcification of the coronary artery increases with ageing in the same degree in men as in women, and that its prevalence is increased in the presence of coronary heart disease. Specifically, the study showed that in people over the age of 65, the presence of calcification in the coronary artery (coronary artery calcium [CAC]) was detected in 60% of healthy subjects and about 95% in patients with coronary heart disease. This study also highlighted that the incidence of calcification under the age of 65 was lower in women than in men.13
To date, many studies have examined the relationship between CAC and conventional cardiovascular risk factors. Virtually all studies agree with the observations published in Lancet in 1964, where age and male gender are key predictive factors. Thus, women delay the development and extension of CAC by 10 years. However, studies do not show a consistent agreement of the impact of the other cardiovascular risk factors on the development of coronary calcification. In this regard, Oei et al.14 reported that 30% of men and 15% of women without cardiovascular risk factors had extensive coronary calcification, and Silverman et al.15 observed that approximately 35% of subjects with ≥3 risk factors have zero CAC. Together, these observations suggest that the presence of cardiovascular risk factors in itself is not a reliable indicator of CAC, and vice versa. However, studies have validated that the presence of a high CAC burden, even among individuals without cardiovascular risk factors, is associated with a high rate of clinical events, while the absence of CAC, even among those with many cardiovascular risk factors, is associated with a low rate of events.14
Characteristics of calcium (volume and density) on the risk of cardiovascular eventsIn 2008, the Multi-Ethnic Study of Atherosclerosis MESA prospectively analysed, in 6722 participants aged between 45 and 84 years and of multiethnic composition 39% non-Hispanic whites, 12% Chinese Americans, 28% African Americans and 22% Hispanic Americans), the influence of the volume of CAC on cardiovascular risk. All participants were free of cardiovascular disease at baseline and were followed-up for 10 years. The study data showed a direct relationship between the volume of CAC and the risk of suffering from coronary heart disease, and consolidated CAC as a strong predictor of future cardiovascular events.16 In line with these observations, a meta-analysis of 30 prospective cohort studies showed in 2009 that the presence of vascular calcification was associated with an increased risk of cardiovascular death, coronary events and cerebral infarction, as well as all-cause mortality.17 Moreover, a recent study by MESA has just shown that CAC is also strongly and gradually associated with an increased risk of cardiovascular events associated with atherosclerotic disease, regardless of standard risk factors, and similarly by age and gender.18 However, it is important to note that a sub-study of the MESA trial conducted on 3398 subjects found that, unlike the volume, the density of CAC was inversely and significantly associated with an increased risk of coronary and cardiovascular disease regardless of CAC volume.19 These findings support the concept that larger and denser calcifications can stabilise atherosclerotic plaques,20,21 while smaller and scattered calcifications appear to contribute to plaque destabilization.22,23 In fact, Krishnamoorthy et al.24 reported that irregular calcification detected by computed tomography is probably associated with plaque rupture. Therefore, it seems more precise to emphasise that the morphology of calcification is what directly determines the associated cardiovascular risk.
Regarding the power of the quantification of CAC in the stratification of cardiovascular risk, the latest clinical recommendations of the ACC/AHA published in 2013 for the use of CAC quantification in asymptomatic patients is limited to those with an intermediate estimated risk in order to improve cardiovascular risk assessment.25 However, they qualify it with a relatively low recommendation level (IIb). On the other hand, the MESA study published in 2016 concluded that if absence of coronary calcium is detected (calcium score = 0 in a coronary computed tomography) in a patient this makes it possible to rule out coronary disease with a very high probability and reclassify the cardiovascular risk to lower levels.26
Osteoporosis and vascular calcificationFor many years, the coincidence in time between the demineralisation of bone tissue, characteristic of osteoporosis, and the arterial calcification that accompanies atherosclerosis was considered coincidental because they were two chronic diseases that increased markedly with age. However, more recent epidemiological studies have shown that there is a close relationship between bone mass loss and vascular calcification, which is independent of age.27 Several studies in the 1990s revealed an association between osteoporosis and vascular calcification, particularly among women.4 Framingham's study showed that women with a greater magnitude of bone loss had a more severe progression of abdominal aortic calcification after correcting for age and other common aetiopathological factors.28 This association was not as manifested in men, which evidenced the crucial role of oestrogen in the underlying pathophysiology. Subsequently, it has been observed that this association between osteoporosis and vascular calcification is also detected at the coronary, carotid and peripheral levels.29,30 In fact, these two conditions share, in addition to ageing, a multitude of risk factors such as dyslipidaemia, hypertension, type 2 diabetes, smoking, alcohol consumption, physical activity and the menopause.4 Although being overweight is considered a cardiovascular risk factor, in osteoporosis, individuals with low weight have a higher risk of fracture.31 It should be noted that this paradox has also been attributed to a specific tissue response to chronic inflammation processes.32 Specifically, as discussed in the previous sections, while a permanent inflammatory response promotes bone formation at the vascular level,33 on the contrary it induces decalcification in hard tissues.34
In addition to sharing risk factors, more recently pathophysiological mechanisms in common in both entities have been suggested, among which we would highlight the intercellular protein system RANK/RANKL/OPG, as well as the Wnt signalling pathway.
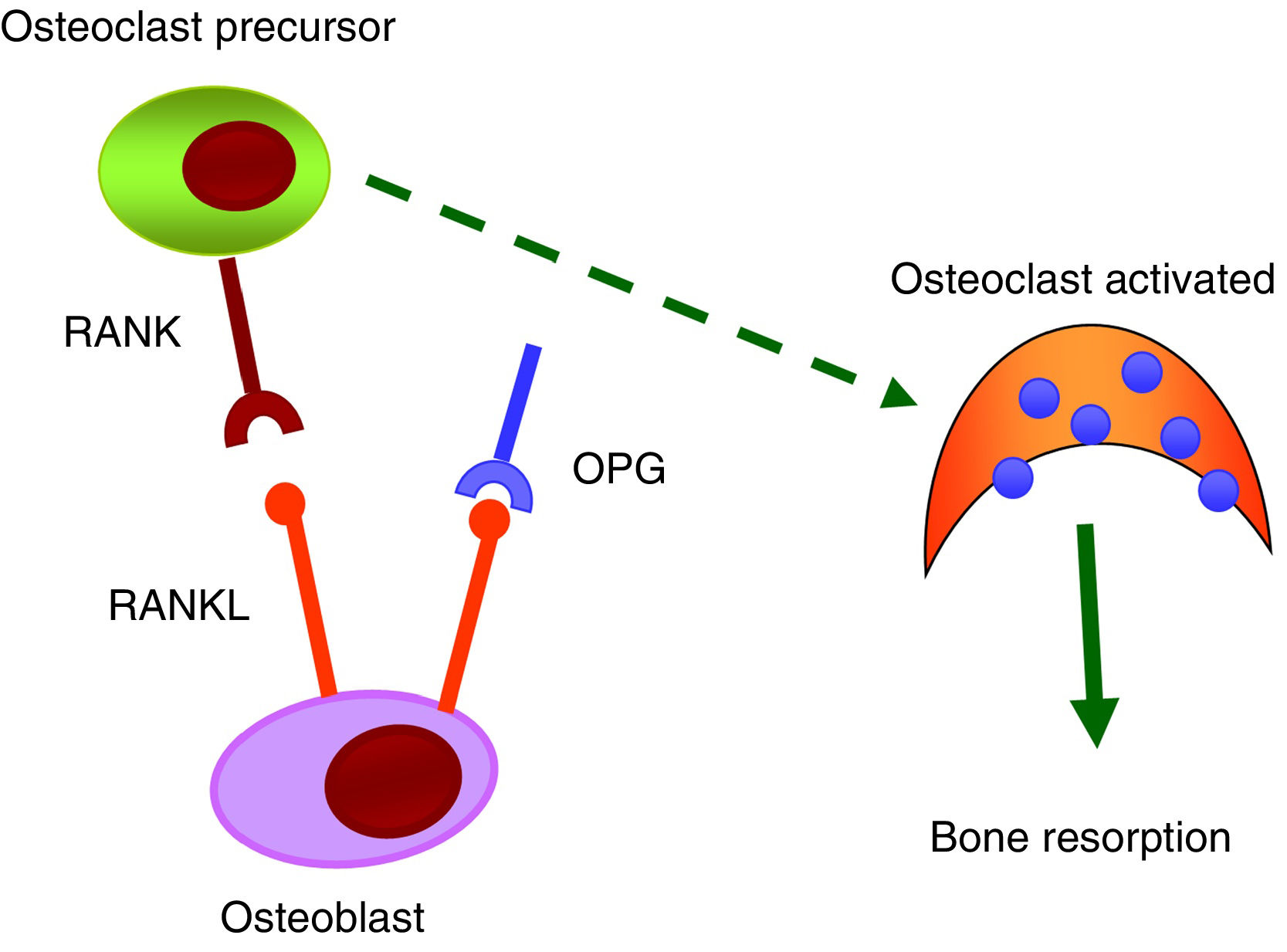
RANK/RANKL/OPG systemThe RANK/RANKL/OPG system is responsible for the activation and differentiation of bone cells.35 All its components belong to the TNF family. RANK is a transmembrane protein that is expressed in the membrane of osteoclasts, but also on the surface of B and T lymphocytes, fibroblasts and dendritic cells. RANKL is a transmembrane protein expressed by osteoblasts and mesenchymal cells. The binding of RANKL to its natural RANK receptor promotes the activation of the NF-κB intracellular signalling pathway and, as a result, generates differentiation of preosteoclasts in mature osteoclasts, facilitating bone resorption. OPG, a protein secreted by osteoblasts and bone marrow stromal cells, is considered to be protective of bone tissue by preventing RANK/RANKL binding and inhibiting the differentiation of preosteoclasts into mature osteoclasts (Fig. 3).

Numerous scientific evidence relates the RANK/RANKL/OPG system with vascular calcifications.36 In the early stages of the vascular calcification process, vascular smooth muscle cells (VSMC) undergo a change in their phenotype and begin to express osteogenic markers that would allow mineralisation of the extracellular matrix. It has been observed that OPG as well as RANKL and RANK proteins are present in atherosclerotic plaques and in valvular disease, and that their level of expression varies depending on the stage of the disease.36,37 OPG seems to be protective against vascular calcification. Studies carried out in knockout mice for osteoprotegerin (OPG_/_) have shown that they developed spontaneous arterial calcification.38,39 Other studies have also shown that RANKL increases the calcification of VSMC by binding to RANK. In short, it is suggested that the RANK/RANKL bond may be important to promoting vascular calcification, while OPG inhibits it.40
Wnt-β-catenin signalling pathwayWnt signalling pathways play an important role in the development and maintenance of many organs and tissues,41 including bone. However, the role of Wnt signalling pathways in the vessel wall is still poorly understood.42
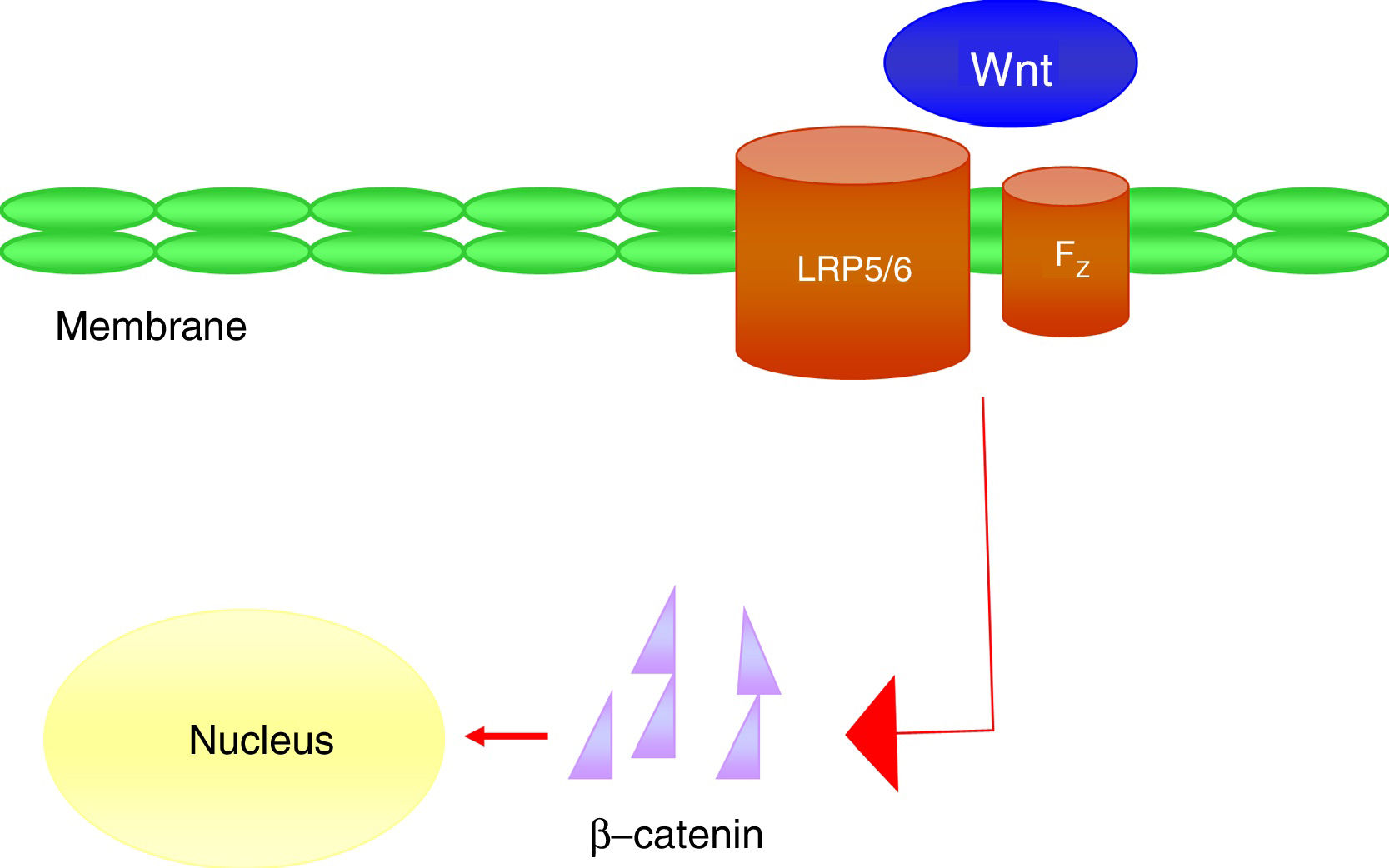
It is a group of signal transduction pathways formed by proteins that transfer signals from the outside of a cell through the receiving surface of said cell to its interior. Wnt proteins signal through various pathways to regulate cell growth, differentiation, function and death. In particular, the Wnt-β-catenin or canonical path seems to be important for bone biology, because it is crucial in osteoblast differentiation and bone formation.43 Wnt-β-catenin signalling pathway activity depends on the cytoplasmic concentration of β-catenin and the proper functioning of the co-receptor complex formed by a protein related to the LDL receptor (LRP5/6) and a frizzled (Fz) receptor, which involves the activation of gene transcription mechanisms in the nucleus mediated by β-catenin, and that in turn will regulate the expression of genes related to the differentiation or function of the osteoblast (Fig. 4). Sclerostin and Dkk1 protein (Dickkopf-1) are the main antagonists of the Wnt pathway when binding to membrane receptors (mainly LRP-5 and -6) and inhibit the activation of said pathway and, consequently, osteoblastic activity.

At the vascular level, it has been observed in elderly women that plasma levels of Dkk1, an inhibitor of the Wnt pathway, are inversely associated with the presence of arterial calcifications.44 These data support the concept that Wnt-β-catenin signalling is an important regulator of vascular mineral metabolism. Due to the similarities between bone formation and calcification, it is considered that the inactivation of the Wnt pathway could attenuate the process of vascular calcification.
Influence of the treatment of cardiovascular risk factors on bone metabolismLipid-lowering agentsRecently published articles suggest that the use of statins is associated with an increase in bone mineral density (BMD) and a reduction in the risk of fracture, even after adjusting for age, sex and comorbidities.45 This protective effect improves in parallel with the accumulated doses of statins or with the exposure time and depends on their potency.46 High-potency statins (atorvastatin and rosuvastatin) and moderate-potency statins (simvastatin) are more effective in reducing the new development of osteoporosis; however, no association has been observed between recent-onset osteoporosis and low-potency statins (lovastatin, pravastatin and fluvastatin). This positive effect of statins on bone is attributed in part to their ability to inhibit osteoclastogenesis, because they increase the expression of OPG mRNA, which prevents RANK/RANKL binding, and thus the differentiation of preosteoclasts to mature osteoclasts.47 Data on bone effects of peroxisome-proliferator-activated receptor agonists α (PPARα]), including fenofibrate, are very limited and contradictory. In animal models, it has been observed that they decrease the expression of osteocalcin in osteoblasts, which negatively affects bone structure and resistance48; but it has also been suggested that they can prevent bone loss, by decreasing the formation of osteoclasts in cultures derived from mouse bone marrow.49 There are currently no data available in the literature regarding the effect of proprotein convertase subtilisin/kexin type9 (PSCK9) inhibitors on bone.
Hypoglycaemic agentsPatients with diabetes have a higher risk of fractures, not only by decreasing BMD, but also by altering bone quality, especially in type 2 diabetes mellitus (DM2).50 Both insulin deficiency and hyperglycaemia deteriorate bone. Therefore, it would be expected that drugs that improve diabetes control could prevent changes in bone tissue, although this does not seem to be the case with all drugs.
Regarding oral antidiabetic drugs, a potential positive effect of metformin on bone has been observed in some studies, showing a fracture risk of up to 20%,51 unlike the A Diabetes Outcome Progression Trial (ADOPT) study, in which no beneficial effect of metformin on fracture risk was demonstrated.52 However, the in vitro models suggest that metformin can have a direct osteogenic effect by promoting differentiation of the osteoblast lineage from multipotent stromal cells derived from adipose tissue, which favours bone formation.53 The use of sulphonylureas is considered neutral or minimal for human bone, as there is no clear correlation between the use of these agents and the incidence of fractures.54 Special attention should be given to the treatment of diabetes with thiazolidinediones or glitazones, PPARγ agonists, particularly in older women, because an increased risk of fracture with this class of drugs has been demonstrated in randomised clinical trials.55,56 It is considered to be a class effect, and the risk factors related to the increase in fractures in glitazone users are female sex, increasing age, pre-existing conditions (comorbidities, glucocorticoid use, smoking and history of previous fractures) and duration of treatment.57 PPARγ agonists favour the differentiation of the mesenchymal stem cell towards the adipocyte to the detriment of osteoblastogenesis.49 In addition, it has been described that they stimulate the production of RANKL, a fact that favours osteoclastogenesis.58 In relation to incretins and dipeptidyl peptidase-4 (DPP-4), in a meta-analysis of randomised clinical trials it was observed that incretins did not modify the risk of bone fracture in patients with DM2,59 while it is believed that DPP-4 could have a positive effect on bone metabolism and could reduce the risk of fracture,60 although these results have not been confirmed in another subsequent meta-analysis.61 Although in vitro models and models in animals have shown that they inhibit bone resorption, while promoting bone formation and quality,57 clinical trials are needed to clarify if there are similar and clinically relevant effects in humans. Regarding sodium–glucose cotransporter 2 (SGTL2) inhibitors, also known as gliflozins, the available data are controversial. According to a meta-analysis that included 38 randomised clinical trials, no differences in fracture risk were observed between patients treated with SGTL2 (dapagliflozin, canagliflozin or empagliflozin) and controls.62 However, in the CANagliflozin cardioVascular Assessment Study (CANVAS) study an increased risk of fractures was observed with canagliflozin treatment.63 Insulin use has been associated with an increase in fractures,54,64 partly attributed to a greater propensity to present episodes of hypoglycaemia, and consequently to an increase in the risk of falls.65 It has also been observed in animal models that impaired bone blood flow stimulated by insulin is associated with harmful changes in trabecular bone microarchitecture and cortical biomechanical properties in DM2. This suggests that vascular dysfunction could play a causal role in diabetic bone fragility.66
AntihypertensivesThere are data in the literature that confirm that thiazide diuretics have a positive effect on bone mass density and in reducing the risk of fracture.67 Traditionally, these effects have been attributed to the increase in renal calcium reabsorption that occurs as a result of the inhibition of thiazide-sensitive sodium chloride co-transporter in the distal tubule.68 Subsequently, it has been observed in animal models that they stimulate the production of osteoblast differentiation markers related to Runx2 and OPN.69 Loop diuretics have been associated with a negative effect on bone tissue,70 while spironolactone seems to have the potential to preserve BMD in the context of primary or secondary hyperaldosteronism. Regarding the drugs that block the renin-angiotensin system, angiotensin converting enzyme (ACE) inhibitors and angiotensin II receptor blocking drugs (ARA-II), are considered to have a beneficial effect on the bone, when a slight decrease in the risk of fracture is observed,71 especially with ARA-II.72 Even so, studies conducted so far are scarce and have limitations, so there is no conclusive consensus on this. There are few studies that evaluate the effect of beta blockers on bone. In animal models, it has been shown to inhibit bone resorption by inhibiting RANKL-mediated osteoclastogenesis.73 A recent meta-analysis showed a 15% fracture risk reduction in patients treated with beta blockers, and cardioselective agents were the most effective.74 Regarding calcium channel blockers, the data available to date have not shown any significant effect of these drugs on bone metabolism.75 No data are available regarding the effect of α-blockers on bone, probably because they are the least used antihypertensives. However, it is believed that they can indirectly increase the risk of femoral fracture, due to their vasodilator effect, which induces low blood pressure and, consequently, increases the risk of falls and fractures.
Influence of osteoporosis treatments on vascular calcifications or cardiovascular riskCalcium supplementsThere is controversy about the role of calcium supplements in vascular calcifications and in atherosclerotic processes. According to data obtained from the Framingham cohort, the intake of baseline calcium in the diet and supplements does not appear to increase or decrease vascular calcification after four years.76 However, in a longitudinal cohort study that included 61,433 women with a 19-year follow-up, a U-shaped relationship with total calcium intake was observed, so that calcium intake >1400 mg/day was associated with higher overall mortality, including that of cardiovascular cause. It should be stressed that this relationship was more pronounced with the use of supplements than with calcium in the diet.77 The relationship between calcium intake and cardiovascular risk is complex and seems to depend on the dose and the source of calcium intake. Even so, based on the available evidence, the recommended dose of 1000–1200 mg/day does not seem harmful at the cardiovascular level, especially if the contribution comes from the diet.78
Selective oestrogen receptor modulatorsThe role of selective oestrogen receptor modulators (SERM) on vascular calcifications is unknown. In in vitro models it has been observed that estradiol and raloxifene affect OPG secretion of endothelial cells, which may suggest a modifying role in the pathogenesis of vascular calcification in postmenopausal women.20,21,79
Antiresorptive drugsBisphosphonates appear to have the potential to reduce the progression of vascular calcifications, depending on the type, potency, dosage and route of administration. This effect is more modest with nitrogen or amino-bisphosphonates administered orally.80 Denosumab, a monoclonal antibody that acts as a RANK ligand (RANKL) inhibitor, has not been shown to have an effect on the progression of aortic calcification or on the incidence of cardiovascular events relative to placebo.81
Osteoform drugsThere is hardly any data in the literature regarding teriparatide (analogue of human parathyroid hormone) and vascular calcifications. In animal models, it has been observed that teriparatide did not affect the calcium content of cardiovascular deposits.82
ConclusionsThe available evidence indicates the interaction between bone mass loss and vascular calcifications. The nature of this relationship is very complex and its clinical importance is still unclear. Both processes share many of the known risk factors for cardiovascular disease, and pathophysiological mechanisms involved in both entities have been suggested, among which we would highlight the RANKL/RANK/OPG system and the Wnt signalling pathway. According to this holistic view, patients with osteoporosis could benefit from an evaluation of their cardiovascular risk, as well as patients with cardiovascular disease from an evaluation of their risk of bone fracture. The mechanisms linked in both entities should be considered in clinical decisions, since osteoporosis treatments may have unforeseen effects on arterial calcification, and vice versa. In particular, it is considered that statins and thiazides may have a beneficial effect on bone, as well as bisphosphonates on arterial calcifications. On the contrary, the use of PPARγ agonists (thiazolidinediones or glitazones) is discouraged, as an increased risk of fracture was shown in randomised clinical trials, especially in elderly women.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: García-Gómez MC, Vilahur G. Osteoporosis y calcificación vascular: un escenario compartido. Clin Investig Arterioscler. 2019. https://doi.org/10.1016/j.arteri.2019.03.008