




La craneosinostosis conlleva la fusión precoz de una o varias suturas craneales, generando un desarrollo anormal de la bóveda craneal, asociado hasta en un 20% a síndromes genéticos. Conllevan buen pronóstico si son corregidas en los primeros meses de vida, disminuyendo así secuelas estéticas y cognitivas. Presentamos, basándonos en un caso clínico, el diagnóstico prenatal, manejo obstétrico y la mala evolución neonatal de un feto afecto de síndrome de Crouzon.
Craniosynostosis is defined as early fusion of one or more cranial sutures, resulting in abnormal development of the skull, associated with genetic syndromes in up to 20%. The prognosis is good if the cranial sutures are corrected in the first months of life, reducing esthetic and cognitive sequels. We present the prenatal diagnosis, obstetric management and poor neonatal outcome of a fetus with Crouzon syndrome.
Las anomalías en la morfología de la calota craneal íntegra, derivadas de una fusión precoz de una o varias suturas craneales, se conoce como craneosinostosis1. Este hecho puede producirse prenatalmente o en los primeros años de vida, generando no solo malformaciones estéticas, sino también, sin corrección quirúrgica, defectos visuales y auditivos y en casos más graves hipertensión craneal con repercusiones cognitivas2.
Su gravedad dependerá del momento de aparición (peor pronóstico cuando sean más precoces) y del número de suturas afectadas (peor cuanto mayor sea el número). A su vez, si asocian síndromes genéticos (Crouzon, Alpert, Pfeiffer, Saethre-Chotzen…) suelen presentar mayor morbilidad.
Estas alteraciones si son aisladas y corregidas, en general, conllevan un buen pronóstico. Presentamos el caso de un feto afecto de síndrome de Crouzon, con craneosinostosis severa visualizada prenatalmente, con una muy mala evolución posnatal.
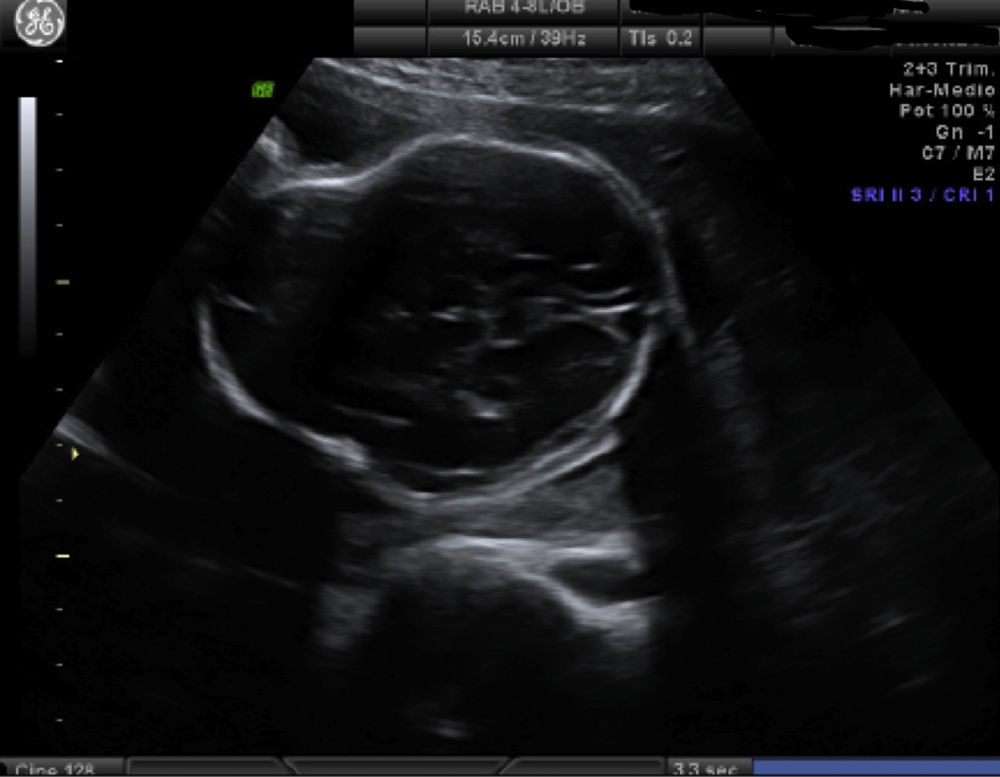
Caso clínicoPrimigesta de 35 años, sin antecedentes de interés. En el estudio ecográfico morfológico en la semana 20 se observa una cabeza en «forma de trébol» con fusión de las suturas coronal, lamboidea y sagital (fig. 1). Sin anomalías en fosa posterior, tubo neural y extremidades; es un feto varón. Ante la sospecha de craneosinostosis, se amplía el estudio con resonancia magnética, describiendo una dolicocefalia en plano sagital con abombamiento frontal y una ligera hipoplasia del macizo facial con proptosis ocular. El estudio de parénquima cerebral y fosa posterior no presenta alteración. La amniocentesis genética evidencia un feto 46-XY, detectándose mutación del gen FGFR3, quedando diagnosticado de síndrome de Crouzon.
.")
Se informa a la pareja del diagnóstico definitivo, de las posibles complicaciones tanto a corto como a largo plazo, por parte del Servicio de Obstetricia, Neonatología y Consejo Genético. Tras ser entendidas y aceptadas, se decide controles ambulatorios con ecografía cada 2 semanas.
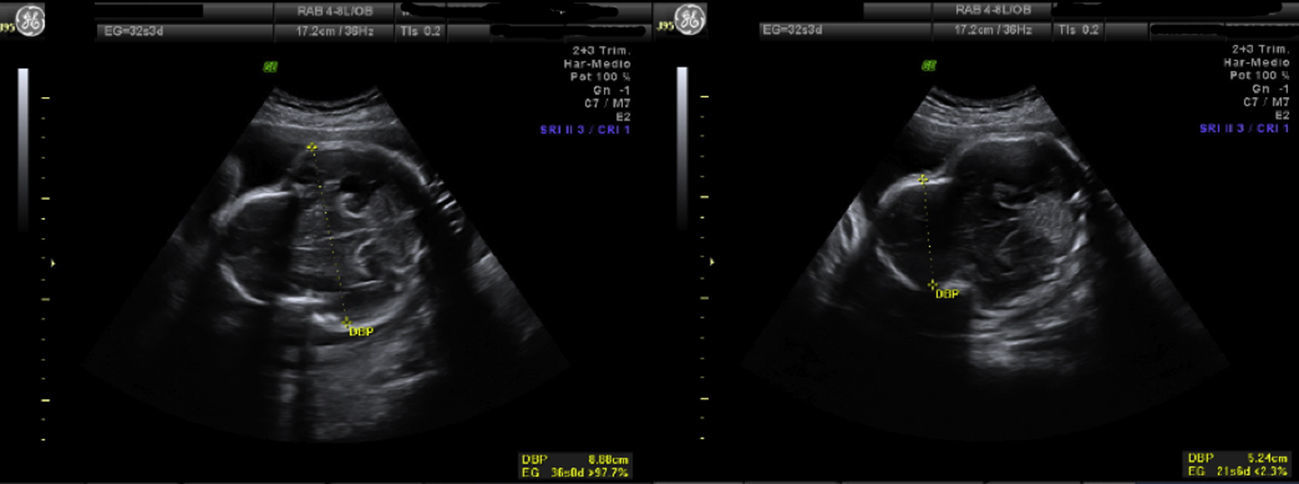
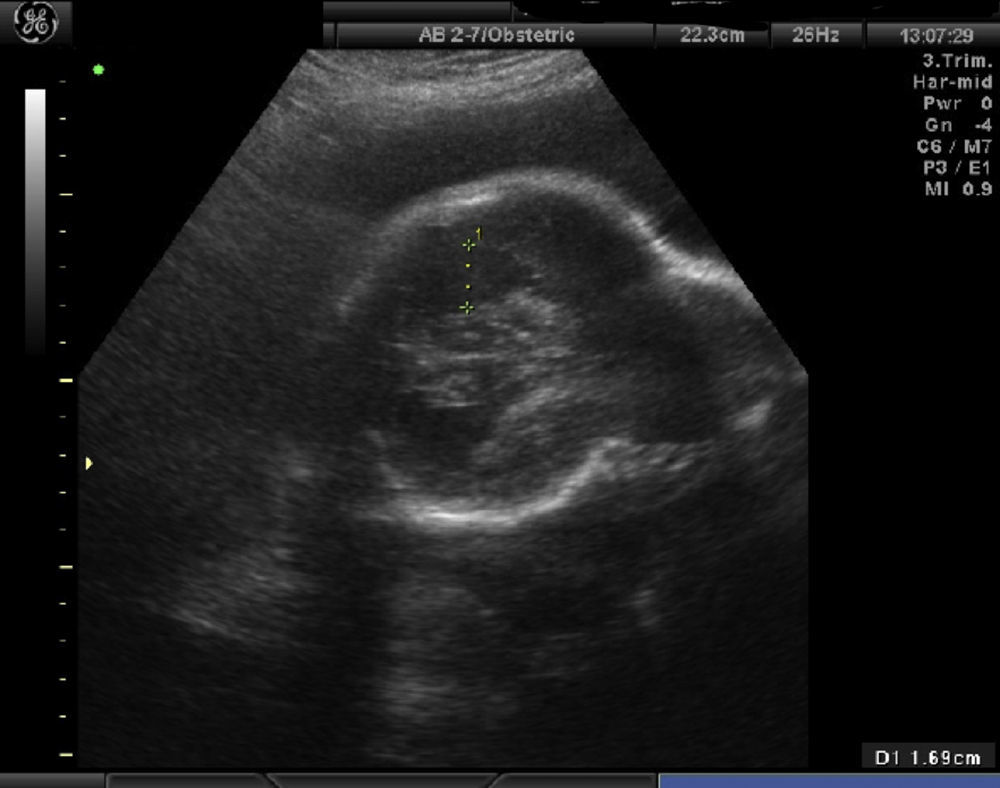
Los controles ecográficos posteriores fueron normales hasta la semana 32. Se evidencia una oxicefalia muy marcada, delimitando claramente una parte anterior en la morfología ósea craneal (con escaso desarrollo) de una parte posterior (fig. 2), siendo el primer control ecográfico en el que aparece polihidramnios (laguna máxima de 10,8cm); el interior de polo cefálico se mantiene normal. El siguiente estudio, en la semana 35, se observa un feto en cefálica con PFE de 2.525g, con un importante polihidramnios (laguna máxima de 16,5cm), apareciendo ventriculomegalia en asta posterior (fig. 3).
 y del anterior (derecha).")

La paciente consulta en la semana 36 por sensación de mojada, confirmándose rotura prematura de membranas, con una presentación cefálica. Dada la imposibilidad de parto vía vaginal (DBP de 99,6mm) se finaliza gestación mediante cesárea bajo cobertura antibiótica, obteniéndose un recién nacido varón de 2.930g con IA: 2/8, pH arterial de cordón umbilical de 7,36. Se evidencia morfología craneal en trébol con protrusión frontal, hipertelorismo y exoftalmos bilateral; el resto de anatomía macroscópica sin lesiones.
El neonato fue intubado e ingresó en UCIN. En los primeros días de vida requirió dilatación mecánica y tutorización de ambas fosas nasales por obstrucción aérea con coanas permeables; mediante técnicas de imagen se evidenció una estenosis simétrica en ambas fosas nasales en su tercio medio-anterior debido a engrosamiento óseo procedente del hueso vómer. En la TAC craneofacial se evidencia una hipoplasia malar y mandibular ligera, con disostosis craneal ya descrita (abombamiento parietal con protrusión frontal debido a la sinostosis del tercio anterior de la sutura sagital, ambas coronales y lamboidea).
A los 30 días de ingreso, tras empeoramiento clínico, se detecta extenso daño isquémico-hemorrágico en sustancia blanca frontal con importante ventriculomegalia, todo ello justificado por un mal drenaje venoso cerebral consecuencia de la malformación ósea, dada la presencia de estrechez en agujeros yugulares y la aparición de un importante sistema venoso colateral en el estudio por resonancia magnética craneal.
Tras ello se realiza derivación ventriculoperitoneal, con disminución ligera en controles posteriores. No puede realizarse tratamiento quirúrgico de la craneosinostosis dada la inestabilidad clínica del paciente. En los controles por técnica de imagen persiste leucomalacia severa con múltiples lesiones compatibles con lesiones hemorrágicas sobre matriz germinal derecha.
Tras un nuevo empeoramiento del paciente y valorando su pronóstico vital con la muy mala evolución clínica presentada, se decide de forma conjunta con los padres limitación de esfuerzo terapéutico, muriendo a los 63 días de vida.
DiscusiónLa craneosinostosis resulta de una fusión prematura de una o más suturas craneales, conllevando una deformidad ósea que precisa de reconstrucción quirúrgica, para evitar la anormalidad de la bóveda craneal y los daños en el desarrollo neurológico que pueden acontecer en estos pacientes. Su incidencia se estima en 1 por cada 2.000-2.500 nacimientos vivos, siendo lo más común la fusión precoz de la sutura sagital1. Tal y como demuestran Shillito y Matson, en una serie de 519 pacientes, la sutura sagital estaba afectada en el 56% de las ocasiones, siendo la afectación simultánea de varias suturas un 13%3.
Las suturas son definidas como articulaciones fibrosas que mantienen unidos los huesos planos que conforman la bóveda craneal, existiendo 4 suturas mayores, que se cerrarán posnatalmente. La sutura metópica (unión de huesos frontales) se cierra a los 2 años de vida. En cambio, las suturas sagital (unión entre los 2 parietales), lamboidea (unión parietooccipital) y coronal (unión frontoparietal) lo hacen más tarde.
La forma de crecimiento óseo del cráneo en la especie humana tiene una gran importancia. Se realiza mediante el depósito cálcico de nuevo hueso, alrededor de la línea de las suturas permeables, estimulado por el crecimiento y la expansión cerebral. Se sabe que el cerebro humano cuadruplica su volumen en los 2 primeros años de vida, lo que corresponde al 75% del volumen que alcanza un cerebro adulto. El otro 20% se llevará a cabo hasta aproximadamente en los siguientes 18 años, con lo que, de forma fisiológica, se procederá al cierre cuando el cerebro ha alcanzado su volumen máximo, en torno a los 20 años de vida3. Si la sutura se cierra prematuramente, se inhibe el crecimiento a lo largo de la sutura fusionada, existiendo crecimiento óseo en otras direcciones, típicamente compensador perpendicular a la sinostosis, apareciendo la deformidad.
La craneosinostosis puede aparecer por errores idiopáticos en el proceso de osificación o como consecuencia de alteraciones metabólicas (hipertiroidismo, hipofosfatemia), hemoglobinopatías (talasemia, anemia falciforme), compresión de la cabeza fetal intraútero (gestaciones múltiples o útero bicorne) o contacto con teratógenos (ácido retinoico, valproato, warfarina…)4.
Lo más frecuente es la afectación de una única sutura, apareciendo como un defecto aislado y de forma esporádica (hasta en el 80% de las ocasiones); en cambio, la presencia de múltiples suturas fusionadas suele ser debida a síndromes genéticos, conllevando importante anomalías acompañantes. En estos síndromes es típica la herencia autosómica dominante. De hecho, las mutaciones encontradas con mayor frecuencia corresponden a los genes que codifican la expresión del fibroblast growth factor receptor (FGFR), implicado en la formación de tejido mesenquimatoso5.
La clasificación de las craneosinostosis suele realizarse en función del tipo y número de suturas implicadas: cuando solo afecta a una se habla de formas simples; en cambio, cuando afecta 2 o más suturas son formas complejas. También se puede clasificar en función de si forman parte de un síndrome o no. Si la fusión prematura de suturas va asociada a otras alteraciones (esqueléticas, cardíacas, SNC…), hablaremos de craneosinostosis sindrómicas, en la actualidad más de 180 descritas.
El diagnóstico ecográfico conlleva siempre una exploración sistemática y detallada de la morfología ósea craneal y del macizo facial6. El rendimiento diagnóstico aumenta si va asociada a otras anomalías, como en extremidades (sindactilia, polidactilia) o en región facial (hiper/hipotelorismo, labio leporino, exoftalmos). Generalmente, con alteraciones aisladas suele ser difícil su diagnóstico prenatal. Es importante recordar que las suturas se observan en un plano tangencial a la superficie ósea como líneas hipoecogénicas; si a lo largo de su extensión se observa pérdida de dicha hipoecogenicidad es indicativo de sutura fusionada7.
Ante la presencia de anomalías en la morfología craneal fetal en el estudio ecográfico es obligado realizar un cariotipado y estudio cromosómico. Se sabe que anomalías cromosómicas generan alteraciones en la morfología craneal. Clásicamente una braquicefalia con ventriculomegalia orienta hacia la trisomía 21, así como la cabeza en «forma de fresa» hacia una trisomía 18. A su vez, también es necesario un estudio detallado para la detección de otras malformaciones asociadas, buscando síndromes malformativos7. Por ejemplo, el síndrome de Alpert (mutaciones en FGFR2) suele asociar de forma característica sindactilias complejas simétricas en manos y pies.
Respecto a la conducta obstétrica, el seguimiento suele ser normal, supeditado por las anomalías asociadas que puedan acompañar (por ejemplo, craneosinostosis coronales suelen asociar cardiopatías complejas en aproximadamente un 25% de las ocasiones). Es un hecho poco común, no reflejado en la literatura, el desarrollo de polihidramnios en estos pacientes. Si bien es cierto, el desarrollo de estos marcadores ecográficos podrían suponer un signo de alarma en el control de estas gestaciones, asociándose a un peor pronóstico posnatal, tal y como ha ocurrido en el caso presentado.
Las complicaciones que acompañan a las craneosinostosis aparecen posnatalmente y son derivadas de la deformidad craneal, que impide la correcta expansión cerebral. A su vez, el crecimiento óseo anómalo puede generar alteraciones visuales y/o auditivas (debido a la deformación o disminución del volumen óseo), según la sutura afectada. Es frecuente que asocien tasas elevadas de hipertensión intracraneal (HIC), alteraciones en el desarrollo cognitivo y alteraciones en la conducta, si no son corregidas quirúrgicamente8.
Se ha observado en torno a un 15-20% de elevaciones ligeras en la presión intracraneal en niños con fusión de una única sutura, con desarrollo de hidrocefalia en tan solo el 1% de los casos2. Pero este porcentaje alcanza el 12% si hablamos de craneosinostosis sindrómicas, como por ejemplo las asociadas al síndrome de Crouzon. Generalmente la hidrocefalia aparece después del primer año de vida por obstrucción del flujo venoso; si no es tratada, conlleva afectación de la vascularización cerebral, retraso mental, edema de papila, atrofia del nervio óptico y ceguera. En nuestro caso fue muy precoz, dado que desarrolló una importante HIC con hidrocefalia debido a un mal drenaje venoso en los primeros días de vida, siendo muy indicativo el desarrollo de ventriculomegalia en asta posterior, en el último control previo al nacimiento.
Respecto al desarrollo cognitivo, existe una mayor incidencia en problemas de lenguaje, comportamiento y neurodesarrollo en niños afectos de craneosinostosis aisladas9. Existe controversia sobre la causa y su prevención, aunque parece concluirse que una cirugía precoz puede paliar su aparición.
La tendencia actual es un manejo activo lo más precoz posible8. Se suele realizar antes del año de vida, aprovechando la plasticidad ósea y la expansión cerebral. La presencia de HIC obliga de forma inmediata al drenaje. Respecto a la cirugía, suele realizarse suturectomía con plastia craneal, seguida de ortopedia craneal posquirúrgica. Presentan tasas de morbimortalidad bajas (6,8 y 0,8% respectivamente) pero no es una cirugía exenta de riesgos; existe la posibilidad de lesión del contenido orbitario, parénquima cerebral y componente vascular (seno sagital)10.
Para finalizar, el síndrome de Crouzon es una de las craneosinostosis sindrómicas con mejor pronóstico. Generada por mutaciones en el gen FGFR2, es de aparición esporádica. Asocia una sinostosis de las suturas coronal, sagital y lamboidea, generando una braquicefalia, oxicefalia, hipoplasia de la cara, exoftalmos, hipertelorismo y prognatismo. A diferencia del síndrome de Alpert, estos pacientes tienen un coeficiente intelectual normal, y no asocian anomalías en manos/pies. Sí es cierto que asocian alteraciones en la visión hasta en la mitad de los casos11.
ConclusionesEl síndrome de Crouzon suele ser una entidad de buen pronóstico; con correcciones quirúrgicas precoces se consigue evitar el daño sobre el desarrollo neurológico por la dificultad de expansión cerebral, pero debemos ser conscientes de que esta dificultad de espacio puede ocasionar daños más inmediatos secundarios a un mal drenaje de LCR y vascularización cerebral, generando secuelas severas irreparables.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
