




El síndrome de Herlyn-Werner-Wünderlich (HWW) es una anomalía congénita del tracto urogenital, causada por un fallo de la fusión de los conductos müllerianos. Presentamos el caso de una paciente de 32 años, en estudio de infertilidad. La resonancia magnética nos conduce al diagnóstico definitivo con los siguientes hallazgos: útero didelfo con hemivagina obstruida, hematocolpos y agenesia renal izquierda. Se resuelve con resección del tabique vaginal y drenaje del hematocolpos secundario.
Herlyn-Werner-Wünderlich syndrome is a very rare congenital anomaly of the urogenital tract, involving defective fusion of müllerian ducts. We report the case of a 32-year-old woman with infertility. Magnetic resonance imaging findings revealed a didelphic uterus with obstructed hemivagina, hematocolpos and ipsilateral renal agenesis. The case was successfully resolved through resection of the vaginal septum and drainage of the hematocolpos
El síndrome de Herlyn-Werner-Wünderlich (HWW) es una anomalía congénita de los conductos de Müller, causada por un fallo de fusión. Se caracteriza por un útero didelfo asociado a una hemivagina obstruida por un septo transverso y agenesia renal ipsilateral. La incidencia del síndrome de HWW es del 0,6-10% del total de las anomalías müllerianas (2-3%)1,2.
Suele presentarse después de la menarquia con una masa palpable por hematometrocolpos y dolor pélvico progresivo.
El diagnóstico precoz es importante para evitar complicaciones como endometriosis, adherencias en la pelvis, piosalpinx o piocolpos y problemas de fertilidad. El tratamiento de elección es quirúrgico con resección quirúrgica del septo obstruido y drenaje de la hemivagina obstruida2.
Caso clínicoPresentamos el caso de una mujer de 32 años con síndrome pluriglandular autoinmune tipo II, y agenesia renal izquierda como antecedentes personales. AGO: ciclos menstruales abundantes con dismenorrea intermitente y nuligesta.
Acude a consulta por infertilidad. A la exploración se palpa formación quística, no a tensión, en los dos tercios superiores de la vagina, en cara izquierda que drena al tacto material mucinoso con grumos marronáceos no malolientes.
En la ecografía se observan 2 cuerpos uterinos/hemiúteros de aspecto normal con endometrio secretor. Útero derecho completo y en el útero izquierdo no se visualiza cérvix. Aparece formación quística tabicada de contenido ecogénico de 104×70×90mm situado debajo de los cuerpos uterinos compatible con colpometra y ambos ovarios normales.
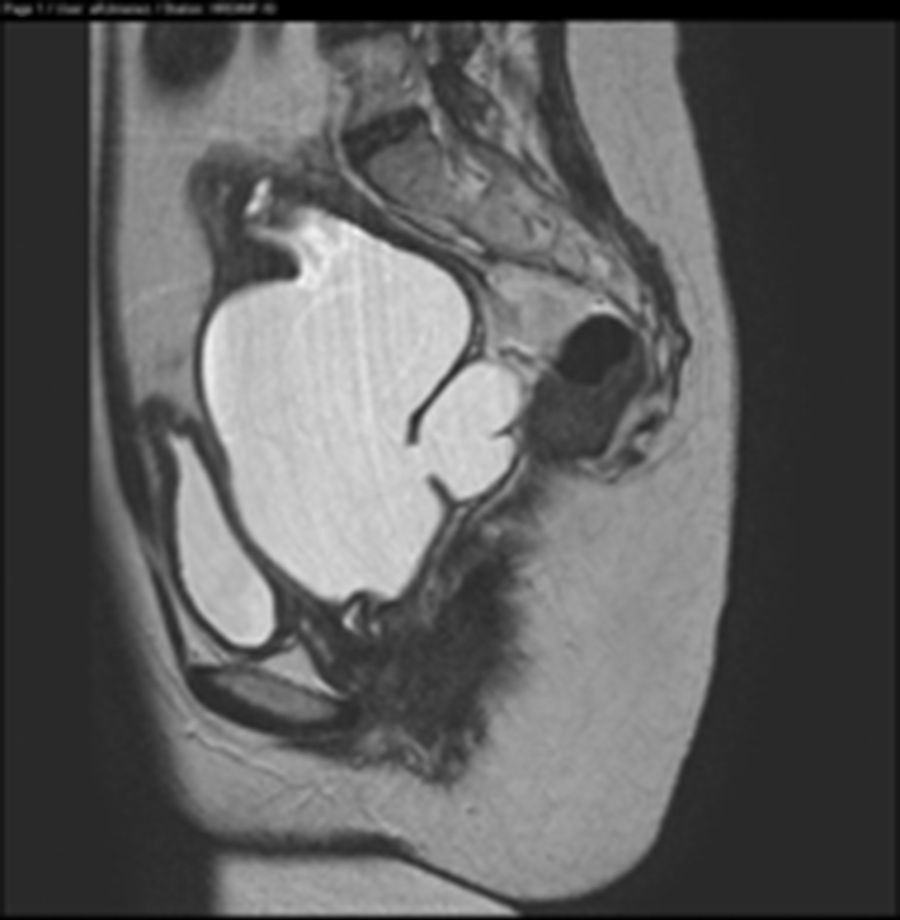
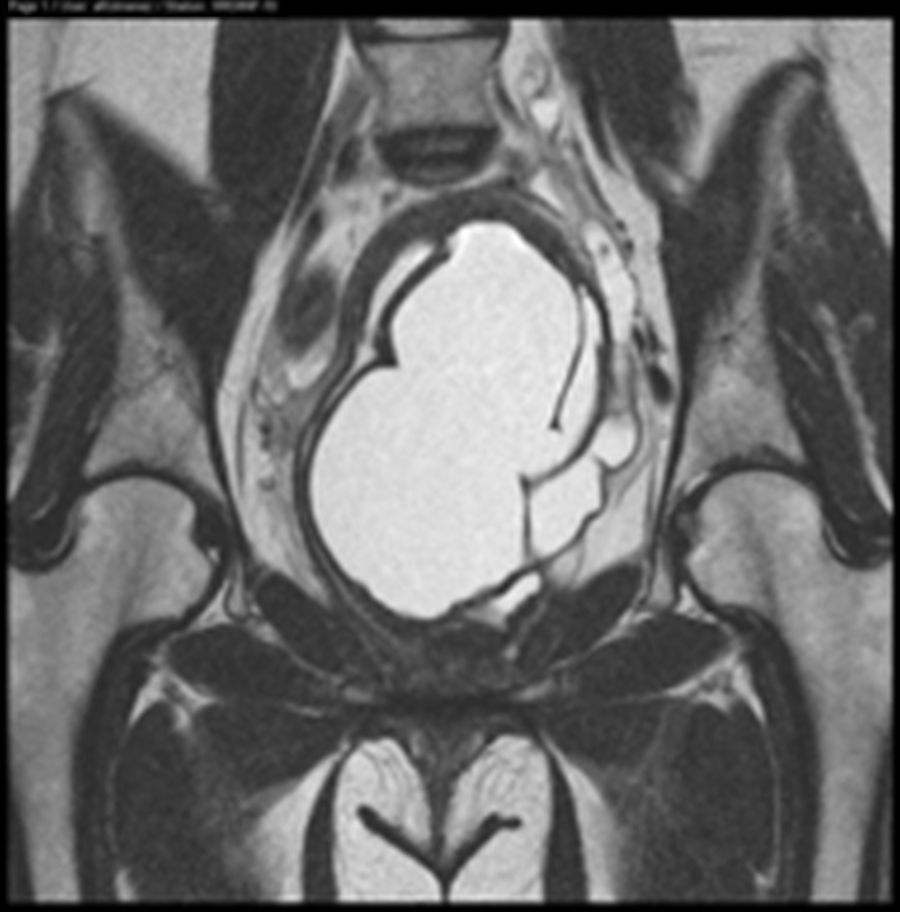
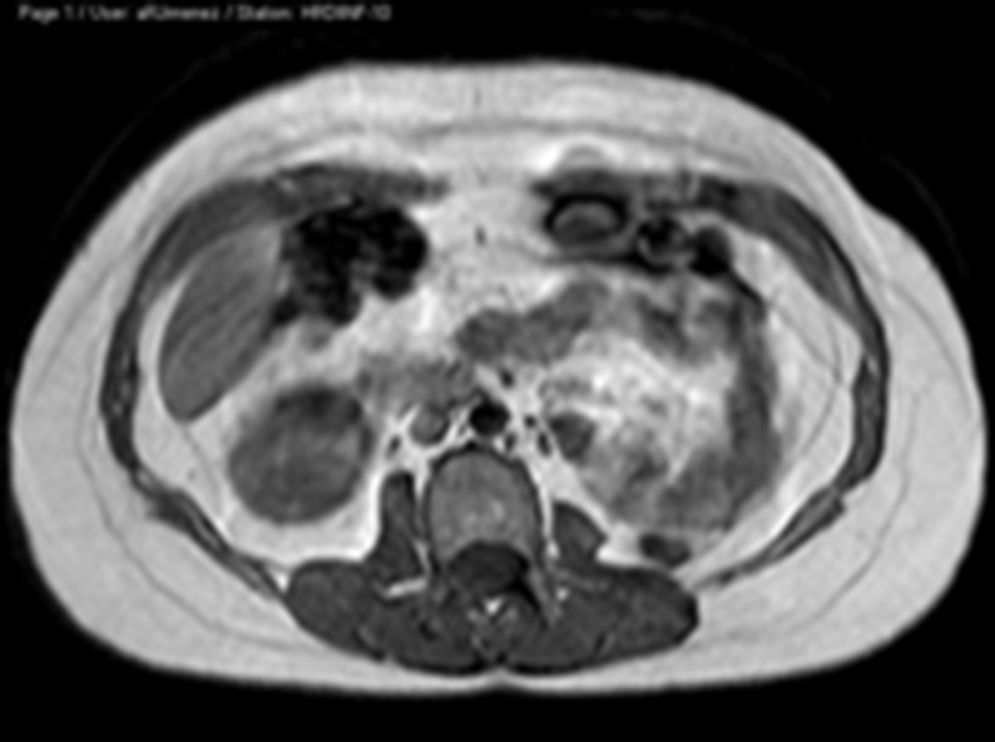
Ante los hallazgos descritos, se solicita una resonancia magnética (RM) que se informa de útero didelfo con 2 cérvix y 2 vaginas, la vagina izquierda ciega, con gran hematocolpos, que por continuidad produce un hidro/hemato-salpinx izquierdo (figs. 1 y 2). Se describe agenesia renal izquierda asociada (fig. 3).



Se propone para resección del tabique vaginal y drenaje del hematocolpos secundario.
Se realiza incisión arciforme de epitelio vaginal en porción más declive de la vagina y punción aspiración de la formación vaginal, saliendo material hemático que se envía para cultivo. Apertura de vagina izquierda, saliendo material hemático abundante. Se realiza histeroscopia de dicha cavidad, encontrando el cérvix izquierdo dilatado. Coagulación y sección de gran fragmento de vagina izquierda con Ligasure®, quedando un pequeño rodete pericervical izquierdo. Postoperatorio sin incidencias y revisiones dentro de la normalidad.
DiscusiónEl síndrome de HWW es una anomalía muy rara dentro de las anomalías de los conductos de Müller. Se produce alrededor de la novena semana de gestación, por un fallo en la fusión de los conductos de Müller verticales y laterales. Su etiología es desconocida y se caracteriza por la tríada de útero didelfo, hemivagina obstruida y agenesia renal unilateral3.
La mayoría se mantienen asintomáticas hasta la menarquia, que empiezan con dismenorrea, dolor pélvico progresivo y masa palpable por el hematocolpos asociado, aunque un 20% se diagnostican a los 20 años, y un 10% más allá de los 30 años como es el caso de nuestra paciente2.
Es importante el diagnóstico precoz para evitar complicaciones como piosalpinx, endometriosis, adherencias y problemas de fertilidad. Aunque muchas veces es difícil por su baja incidencia y por los síntomas tan inespecíficos que presentan.
El diagnóstico de sospecha se realiza con la ecografía, visualizando el hematocolpos y la posible malformación uterina, y se confirma con la RM que presenta una mayor sensibilidad y nos ayuda para la planificación quirúrgica2,4,5.
Se realizará laparoscopia en aquellos casos en que la RM sea dudosa. Aunque la utilidad de esta técnica para el tratamiento quirúrgico no está clara, sí puede ayudar al drenaje del hematocolpos y/o al tratamiento de las complicaciones ya descritas3,5.
Actualmente el tratamiento de elección es quirúrgico, mediante resección y marsupialización de la vagina septada.
El abordaje histeroscópico parece ser la mejor forma de resolución quirúrgica disponible en la actualidad para dichos defectos. El primer caso publicado de corrección resectoscópica de tabique vaginal fue en el 1998 por Tsai et al.6. Posteriormente Cicenelli et al.7 en el 1999 añaden a esta técnica una guía ultrasonográfica durante la cirugía que disminuye el tiempo de intervención y ofrece una mayor seguridad durante la resección8.
Una nueva técnica quirúrgica publicada por Cooper y Merrit9 en el 2010 consiste en el uso de una endoprótesis traqueobronquial para mantener la permeabilidad vaginal después de la excisión del tabique vaginal con el fin de evitar la estenosis vaginal posquirúrgica8.
Según la literatura, la tasa de embarazos en estas pacientes es de alrededor del 87%, un 23% abortan, un 15% presentan partos pretérminos y el 62% restante presentan embarazos a término10.
En nuestro caso, la resección del tabique vaginal y el drenaje del hematocolpos fue exitosa y sin complicaciones, preservando así el potencial reproductivo de la paciente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
