



To implement a selective and sensitive analytical method to quantify morphine in small volumes of plasma by gas-liquid chromatography-mass spectrometry (GC-MS), aimed at post-operatively monitoring the drug.
METHODA gas-liquid chromatographic method with mass detection has been developed to determine morphine concentration in plasma after solid phase extraction. Morphine-d3 was used as an internal standard. Only 0.5 mL of plasma is required for the drug solid-phase extraction in the Bond Elut-Certify®, followed by the quantification of morphine derivative by GC-MS using a linear temperature program, a capillary fused silica column, and helium as the carrier and make-up gas. The method was applied to determine morphine content in plasma samples of four patients during the postoperative period of cardiac surgery. Patient-controlled analgesia with morphine was performed by a venous catheter, and a series of venous blood samples were collected. After the oro-tracheal extubation, morphine plasma levels were monitored for up to 36 hours.
RESULTSThe run time was 16 minutes because morphine and the internal standard were eluted after 8.8 minutes. The GC-MS method had 0.5–1000 ng/mL linearity range (r2=0.9995), 0.1 ng/mL limit of detection, intraday and interday precision equivalent to 1.9% and 6.8%, and 0.1% and 0.8% systematic error (intraday and interday, respectively). The analytical method showed optimal absolute (98%) and relative (100.7%) recoveries. Morphine dose requirements and plasma levels are discussed.
CONCLUSIONThe analytical gas-liquid chromatography-mass spectrometry method is selective and adequate for morphine measurements in plasma for applications in clinical studies.
Opiates are the drugs of choice for short-term treatment of post-surgical and traumatic pain as well as for long-term treatment of severe pain in cancer patients. Because morphine is the most potent painkiller, it is the reference by which analgesics are assessed.1 Particularly for surgical patients, it has been extensively reported that pain and anxiety may cause major discomfort, increase the risk for postoperative complications, and even prolong their hospital residence.2 The use of Patient-Controlled Analgesia (PCA) in hospitals has been a recent practice because of its proven advantages over conventional intramuscular injections. These include improved pain relief, greater patient satisfaction, less sedation, and fewer postoperative complications. Morphine is the most studied and most commonly used intravenous drug for PCA,3 but large or repeated doses can induce prolonged sedation, nausea, vomiting, apathy, reduced physical activity, dysphoria, constipation, hypotension and respiratory depression, which can lead to death.1,4
Although, several analytical methods for morphine detection have been proposed, including radioimmunoassay,5,6 gas chromatography (GC) with nitrogen–phosphorous detection, flame ionization detection and electron capture detection, GC–mass spectrometry (GC–MS) has unsurpassed sensitivity and selectivity.7–15 Many studies have used GC–MS as an analytical tool, but sample preparation is very laborious, and no study has achieved the sensitivity required for pharmacokinetic studies of low doses of morphine.13,15–22
The aim of the present study was to implement a selective and sensitive GC-MS method to determine plasma morphine for application in clinical studies. Typically, plasma concentrations in such studies range from 1 to 20 ng/mL and involve the processing of a large a number of samples.
MATERIALS AND METHODSChemical and reagentsMorphine sulphate pentahydrate (M-8777) was purchased from Sigma Aldrich (MO, USA), and the internal standard Morphine-(N-methyl-d3), used at 10 μg/mL in methanolic solution (M-006 RADIAN®), was obtained from Cerilliant (TX, USA).
Bis(trimethylsilyl)trifluoroacetamide (BSTFA) T-6381, the derivatizing agent, was purchased from Sigma Aldrich (MO, USA). The siliconizing agent, dimethylchlorosilane (DMCS) D-6258, was obtained from Sigma Aldrich (MO, USA). All other solvents and reagents of analytical grade were from Merck (Sao Paulo, Brazil). Water was purified by a Simplicity Milli-Q system from Millipore (Sao Paulo, Brazil). High purity helium was analytical grade 5.0, purity 99.999%, and was obtained from White Martins (Sao Paulo, SP, Brazil).
The solid phase extraction apparatus consisted of a solid phase cartridge Bond Elut-Certify®, 50 mg, 50/PK, Varian-12105030 (CA, USA) attached to a vacuum system Welch Vacuum® (IL, USA ). In addition, a conic Becker model 13222, a Reacti-Vap 18780 and a Reacti-Therm 18870 system, all supplied by Pierce (IL, USA) were used to obtain the morphine derivative.
InstrumentationAnalyses were performed in a Hewlett-Packard 6890 Series GC (LP, USA) equipped with a splitless injector and a 6890 Series mass selective detector (MSD) in EI mode (70 eV) from Hewlett-Packard (LP, USA).
The column was a 30 m, 5% phenyl methyl siloxane capillary column HP-5MS Hewlett-Packard (LP, USA) with an internal diameter of 0.25 mm and a film thickness of 0.25 mm. The morphine and internal standard peak:area ratios were plotted using an HP Deskjet 890C, Hewlett-Packard (Sao Paulo, Brazil).
Chromatographic conditionsHigh purity helium was used as the carrier gas at a constant flow-rate of 0.6 mL/min (constant pressure, 7 psi) and as a make-up gas for the mass detector at a constant flow of 0.2 mL/min. The injector and detector temperatures were maintained at 250°C and 280°C, respectively. The oven temperature was programmed as follows: 150°C for 1 min after injection, increasing to 250°C at 20°C/min, and finally holding at 250°C for 8 min (overall run time 13 min and recovery time 3 min). The MSD was operated in the selected-ion monitoring mode (SIM) using m/z equivalent to 429.0 and 432.0 for the morphine and morphine d3 (internal standard), respectively; each ion had a dwell time of 100 ms.
Preparations of standards and internal controlsStock solutions of morphine were prepared by dissolving the appropriate amount of standard in pure water to yield a final drug concentration of 1 mg/mL, free base. Linearity was investigated by adding an appropriate volume from the stock solutions to drug-free plasma to obtain the following concentrations of morphine in plasma: 1000, 880, 440, 220, 110, 55, 23, 11, 6, 3, 1 and 0.5 ng/mL, with the calibration curve plotted daily by serial dilution in the range 0.5 – 440 ng/mL. Internal quality controls were prepared by diluting the stock solution in drug-free plasma to obtain high (400 ng/mL), medium (200 ng/mL) and low (10 ng/mL) concentrations. 0.5mL aliquots of the standards in plasma were distributed in Eppendorf tubes and stored at −20°C until assay.
Morphine d3, internal standard (IS) and stock solutions were prepared to obtain 1mg/mL in methanol, distributed in Eppendorf tubes, and stored at –20ºC. The working solution of the internal standard was prepared daily from the stock solution to obtain a concentration of 10 μg/mL.
Bis-(trimethylsilyl)-trifluoroacetamide (BSTFA) was sealed in Sigma Aldrich T6381 ampoules (1 mL) and kept in the refrigerator. Dimethylchlorosilane (DMCS), 10%, was prepared by dissolving 1 g with 10 mL of hexane.
Sample extraction procedurePreparation of glasswareGlassware was siliconized by immersion in a 10% solution of dimethylchlorosilane in n-hexane for 60 min, rinsing with methanol and then acetone, and oven drying at 60°C for 30 minutes.
Solid-phase extraction and morphine elutionPlasma aliquots (0.5 mL), 0.5 mL of Tris buffer (pH 9.5) and 25 μL of the working solution (morphine d3/ internal standard) were transferred to sample tubes. Solid phase extraction was performed with Bond Elut Certify (50 mg) cartridges. Each cartridge was first conditioned with 2 mL of methanol, followed by 2 mL of water, both at flow rate of 3 mL/min. After application of the samples at 1 mL/min, the cartridges were washed with 2 mL of water, followed by 1 mL of acetate buffer (50 mM, pH 4), and then 2 mL of methanol. Then, the cartridges were dried with a 10 mL stream of air, and morphine was eluted with 2 mL of a freshly prepared mixture of solvents consisting of dichlormethane, isopropylic alcohol and ammonium hydroxyde (20:5:0.5, v/v). The purified extract eluted from the solid phase extraction was transferred to a previously siliconized derivatization Becker (reaction vial from Pierce, code 13222, IL, USA), and the solvents from the eluate were evaporated to dryness (heating block at 40oC) for later derivatization.
DerivatizationThe purified dried extract was added to 50 μL BSTFA agent. The derivatization was performed at 70°C for 20 minutes in a Reacti-Vap® model 18780, Reacti-Therm® model 18870, both supplied by Pierce (IL, USA). The solution was concentrated under a stream of nitrogen, and the 2 μL aliquots were injected into the GC–MS equipment.
Linearity, calibration curve and calculation proceduresThe nominal value of plasma morphine was plotted as a function of the peak area ratio obtained from morphine and its internal standard (morphine d3) versus the drug concentration. The linearity of the method was determined in triplicate for each concentration ranging from 0.5 to 1000 ng/mL.
The linear regression line and the estimated linear correlation coefficient were used in the calibration curve (equation: y = b + ax, where x is the peak area ratio, a is the slope and b is the intercept). According to the current local regulations for bioanalytical methods, at least five of the total amount of calibrators (0.5, 1, 3, 6, 11, 23, 55, 110, 220 and 440 ng/mL) must be considered for the plot of the daily calibration curve. The daily curve was accepted if at least 7/10 of the internal controls (high, medium and low concentrations analysed in triplicate) presented a systematic error lower than 15%. At least one control at each concentration was required to be within the acceptable range. Once accepted, the calibration curve was used to estimate the drug concentration in samples from patients. The linearity of the method was determined in triplicate at each concentration ranging from 0.5 to 1000 ng/mL.
Accuracy, precision and recoveryThe precision of the method was defined as the degree of agreement among individual tests when the procedure was applied repeatedly and multiple replicates at three different concentrations were analysed and expressed as the coefficient of variation (CV %) from the back-calculated value subtracted from the target value and then divided by the target value, expressed as a percentage. The intra-day precision was evaluated by analysis in triplicate of the high (1000 ng/mL), medium (400 ng/mL) and low (10 ng/mL) concentrations. The inter-day precision was determined by analysis in triplicate of the high, medium and low concentrations of morphine on three different days (n=27).
Accuracy was evaluated by the analysis of multiple replicates in three different concentrations and expressed as a percentage of inaccuracy or systematic error. This parameter was estimated by the value of the mean back-calculated concentrations divided by the theoretical concentrations, expressed as percentage. The intra-day accuracy was evaluated by triplicate analysis of the high (1000 ng/mL), medium (400 ng/mL) and low (10 ng/mL) concentrations. The inter-day inaccuracy was determined by analysis of the high, medium and low concentrations of morphine in triplicate on three different days (n=27).
Absolute recovery of morphine from plasma was estimated by the peak area integrated for morphine in the plasma assayed versus the peak area integrated, after direct injection, expressed as a percentage. The efficiency of the relative recovery of the drug in plasma assayed according to the extraction procedure was estimated by the peak:area ratio obtained for morphine relative to its internal standard.
SpecificityThe specificity of an analytical method is its ability to accurately measure an analyte in the presence of endogenous compounds. Specificity was evaluated by the analysis of drug-free plasma samples (normal, haemolysed, lipemic and icteric sample biological matrices) and applying the analytical procedure.
Limits of detection and quantificationThe limits of detection (LOD) and quantification (LOQ) were determined based on the analysis of ten replicates. The LOQ was defined as the lowest drug plasma concentration of the daily calibration curve that could be determined with an accuracy of 80–120% and precision lower than 20%. LOD had a signal to noise ratio equivalent to 2:1, while the LOQ had a ratio of 10:1.
Stability studyTo determine the stability of morphine in plasma, variations of less than 15% for all the concentrations studied were accepted for use. The stability study consisted of the following assessments:
- •
Spiked blank plasma samples were analysed after three freeze/thaw cycles by GC-MS in the same sequence after cleaning up the plasma samples as detailed above, using two different concentrations of morphine in plasma (10 and 400 ng/mL) analysed in triplicate during three consecutive periods. Data were expressed as percentage of systematic error or inaccuracy. The acceptance criterion for all concentrations studied was set as less than 10% variation.
- •
Conditions for the analysis were determined from samples extracted in triplicate at three concentrations (10, 400 and 1000 ng/mL) and kept at room temperature after derivatization for the maximum time the sample could remain under these condition. The concentration obtained was expressed as a percentage of the nominal value.
- •
Short term stability: Two different morphine concentrations (400 and 10 ng/mL) were used. The controls remained at room temperature for 6 hours, and then were examined by the method described.
- •
Long term stability at −20°C: Three concentrations were used in triplicate for different periods of time to ensure stability throughout the course of the study.
The robustness of the method was determined using slight variations in the flow of gas for drag (± 0.05 mL/min). The study was developed using six replicates, and results were expressed as a function of the coefficient of variation.
Clinical study designSerial blood samples were collected from venous catheters of four patients during the postoperative period after extubation at the following time points: 3, 6, 12, 18, 24 and 36 hrs. Heparinized blood collecting tubes were centrifuged at 1500 g for 15 min. The plasma was separated and stored at −20°C until analysis. The study protocol was approved by the local ethics committees of the two institutions involved: the Department of Pharmacy and the Department of Medicine from the University of Sao Paulo, and written informed consent was obtained from each participant before inclusion in the trial.
Four coronary artery bypass grafting surgery patients were investigated. Characteristics of patients and drug requirements (PCA) in the late postoperative period are shown in table 1.
Demographic and somatic characteristics, drug requirements (PCA) and analgesia after extubation in the late postoperative period
| Patient allocation | Morphine PCA (mg) | Gender | Age (yrs) | Weight (kg) | Height (m) | BMI (kg/m2) |
|---|---|---|---|---|---|---|
| # 04 | 13 | F | 41 | 48.0 | 1.50 | 21.33 |
| # 11 | 10 | M | 63 | 88.0 | 1.72 | 29.75 |
| # 15 | 26 | M | 76 | 67.6 | 1.64 | 25.13 |
| # 30 | 72 | M | 64 | 69.1 | 1.61 | 26.66 |
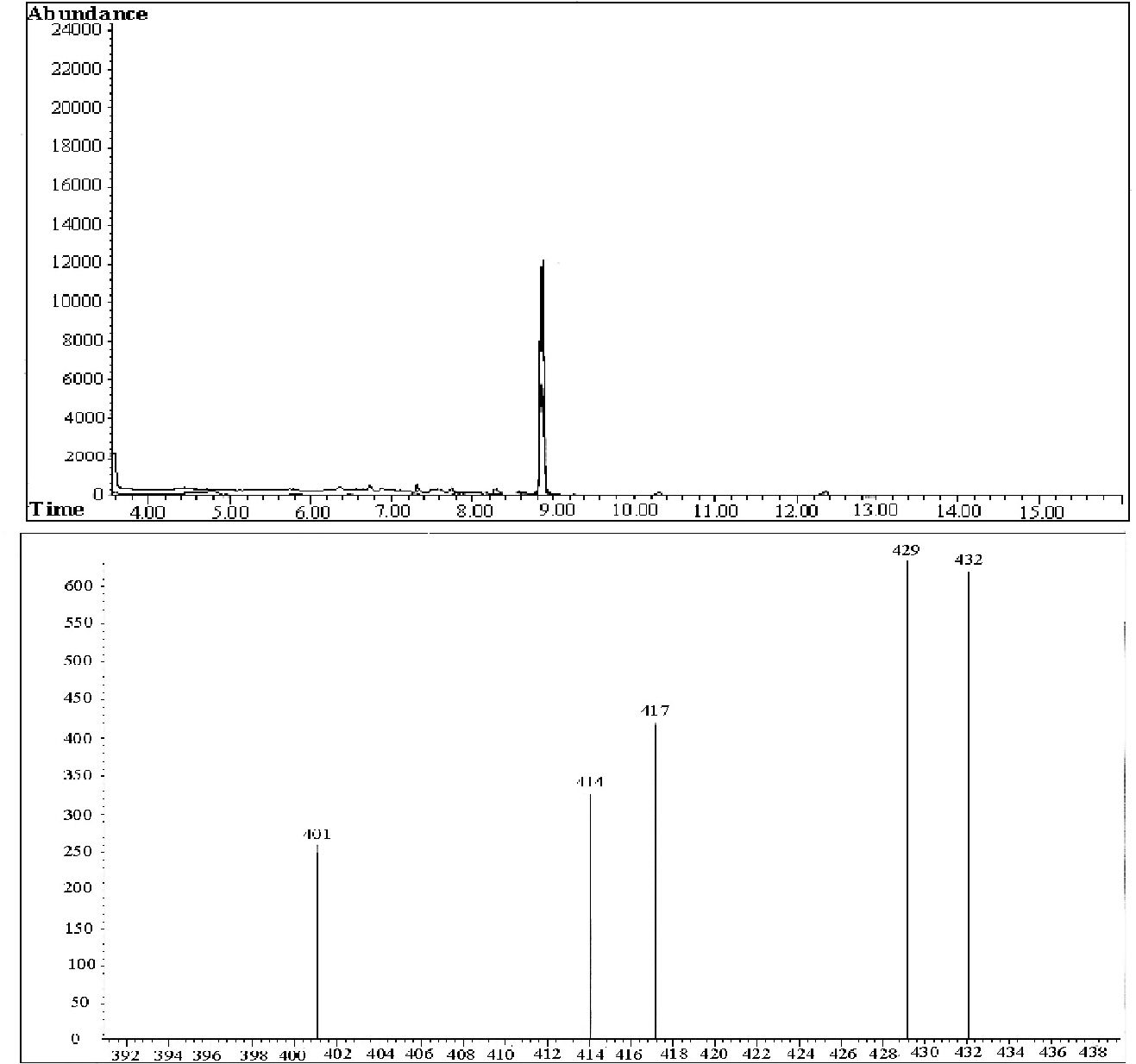
Among the mass/charge (m/z) peaks (401, 414, 417, 429 and 432), 429 and 432 are fingerprints for morphine and the morphine-d3 derivatives, respectively. The retention time was 8.8 minutes for both morphine and morphine-d3. Typical chromatograms of extracts obtained from the determinations of isolated morphine in plasma samples plus the internal standard (morphine d3) and the mass spectrum are shown in Fig. 1. To guarantee the high selectivity obtained, the total time required for each chromatographic run was 16 minutes.
 8.8 minutes. Run 16 minutes. Mass spectrum: mass 429 (morphine); mass 432 (morphine D3).")
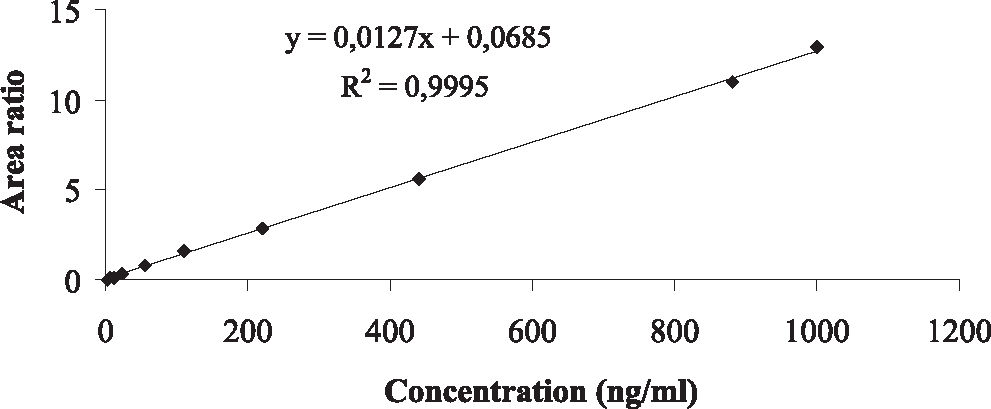
The analysis of morphine content revealed excellent linearity and acceptable accuracy and precision (Table 2). Linearity was expressed by the intercept, and the slope of the linear function as the mean, with the respective standard error of the mean (SEM) as follows: intercept 0.0219 (SEM: 0.002376), slope 0.0128 (SEM:0.000036); also it was considered the linear correlation coefficient (r2 0.9995) over the 0.5 –1000 ng/mL range, Fig. 2. The detection limit was 0.1 ng/mL, and the quantification limit was 0.5 ng/mL based on the analysis of 0.5 mL plasma in 10 replicates.
Confidence limits of analytical method to determine plasma morphine concentration
| Parameter | Unit | Confidence Limits |
|---|---|---|
| Linearity | ng/mL | 0.5 – 1000 |
| Linear regression coefficient | (r2 = 0.9995) | |
| Limit of quantification (n=10) | ng/mL | 0.5 |
| Limit of detection (n=10) | ng/mL | 0.1 |
| Precision (10,200,400ng/mL) | ||
| Intra-day (mean) | (%) | 1.9 |
| Inter-days (mean) | (%) | 6.8 |
| Systematic error (10,200,400ng/mL) | ||
| Intra-days (mean) | (%) | 0.1 |
| Inter-days (mean) | (%) | 0.8 |
| Stability (Thawing cycles)* | (%) | 3.5 |
| Short-term stability * | (%) | −0.5 |
| Long-term stability * | (%) | 5.6 |
| Robustness flow rate (± 0.05 mL / min). | (CV%) | 3.6 |
| Recovery | ||
| Absolute | (%) | 96.4 |
| Relative | (%) | 100.7 |
")
The method showed good sensitivity, linearity and stability, with acceptable accuracy and precision (Table 2). In addition, slight variations in the flow of gas for drag (± 0.05 mL / min) showed a coefficient of variation of 3.6% (Table 2).
Analysis of the short-term stability of morphine (time and conditions of analysis) demonstrated that the drug in the plasma extract submitted to chromatography did not degrade within an 8 h period. The short term stability of morphine had a 0.5% coefficient of variation, and the long term stability of morphine had a 5.6% coefficient of variation (Table 2). Thaw cycles revealed good stability of the drug in biological samples after three consecutive freeze/thaw cycles.
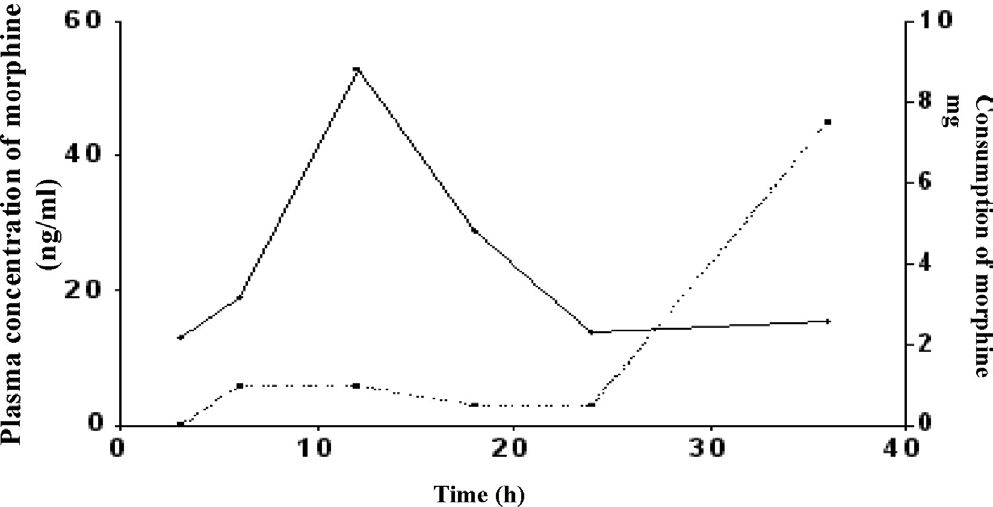
Therapeutic Drug MonitoringDuring PCA device drug bolus delivery in the postoperative period, morphine plasma levels, expressed as medians, were: 13.1 ng/mL (3rd hr), 18.9 ng/mL (6th hr), 53.0 ng/mL (12th hr), 28.9 ng/mL (18th hr), 13.9 ng/mL (24th hr) and 15.4 ng/mL (36th hr) (Fig 3). Additionally, the fraction of morphine PCA consumed at time intervals up to 36 hours was considered to be even, as illustrated in Figure 3. The total amount of the drug delivered up to 36 hours, expressed as a median, was 19.5mg.
 considering the dose of morphine PCA consumed at each interval. Population data expressed as medians: (__) plasma concentration, (–) consumption of morphine up to 36 h.")
Curve of plasma concentrations of morphine obtained as a function of time after extubation in the group of study (n = 4) considering the dose of morphine PCA consumed at each interval. Population data expressed as medians: (__) plasma concentration, (–) consumption of morphine up to 36 h.
Morphine is the analgesic opiate most frequently used for short periods for treatment of post-surgical or trauma pain and even for long-term treatment to control severe pain in patients with cancer. Patient-controlled analgesia is preferred in such cases because the pump device can deliver 1–100 mg of morphine by intermittent boluses.6,12,23–25
Several methods have been proposed to determine the morphine content in biological matrices, mainly in legal medicine for forensic toxicology purposes or in drug abuse control. Only a few methods, however, are adequate for analgesia clinical studies because a large degree of linearity and high sensitivity are required in the follow up of those patients receiving intravenous intermittent boluses of morphine.
Radioimmunoassay (RIA) is quite simple and sensitive but has a serious limitation because an important cross-reaction of the main metabolites, morphine 3-glucuronide (M-3-G) and morphine 6-glucuronide (M-6-G), occurs with the polyclonal antibody.5,6 High performance liquid chromatography using electrochemical, chemoluminescence or fluorescence detection showed low sensitivity compared to RIA. Another limitation in the preparation of the organic extracts is the extensive procedure and long time required to clean up biological matrices. 26–29
Using gas chromatography to determine morphine content in biological matrices in combination with a flame ionisation detector, nitrogen-phosphorus detector or even electron capture detector has problems related to selectivity for morphine at low concentrations.7–15 The advantages to determining morphine content by gas chromatography-mass spectrometry (GC-MS) are the high sensibility and selectivity that facilitated its original application in forensic toxicology and drug abuse control.13,15–22 GC-MS is a suitable method to determine morphine content but requires an internal standard, in general a structural analogue such as nalorphine or morphine-d3.13,15,17–19,30–32 Concerning the purification of the biological matrices, Krogh et al. (1993) described a direct injection of the sample that was diluted, derivatized and injected in an automated CG-MS system. The method presented a serial of operational disadvantages caused by the direct injection of the biological matrix. Particularly for forensic purposes, Ropero-Miller et al. (2002) used the precipitation of proteins from whole blood with methanol, followed by solid phase extraction using cartridges CSDAU 206 (BRISTOL, PA). Meatherall et al. (2005) reported the precipitation of proteins with acetonitrile followed by liquid–liquid extraction using a mixture of chloroform and trifluoromethanol. Lower recoveries resulted from the clean up of biological matrices in both methods; the main disadvantage is that the low end of the sensitivity range reached 10 and 50 ng/mL.31–33 Liquid–liquid extraction of morphine from biological matrices was reported previously, but the required sensitivity for application in clinical trials was not achieved in most cases. 15–18,22,31 Jones et al. (1984) determined the morphine content in urine and tissues for forensic purposes with adequate sensitivity by GC-MS, in spite of the laborious sample clean up process. 34
A solid phase extraction to clean up biological matrices was reported by Drost et al. (1984). These authors proposed the use of Bond Elut C18 cartridges to determine morphine content by GC-MS after derivatization with BSTFA. A low sensitivity, however, was obtained (5 ng/mL), and the linearity was inadequate. Then, a series of different cartridges were investigated to optimise the purification of biological matrices by solid phase extraction, none of which reached the sensitivity and linearity required for clinical studies.12,19–21,35,36
Therefore, in the present study, we designed an analytical procedure to purify the biological matrix by solid phase extraction by applying Bond Elut Certify®, 50mg/3mL (Varian, CA, USA). These cartridges were able to concentrate morphine extracted from the reduced volume of plasma. An optimal absolute recovery (96.4%) was obtained for morphine, and its internal standard (morphined3) guaranteed adequate sensitivity (LOD: 0.1 ng/mL and LOQ:0.5 ng/mL), good precision and accuracy and also a large degree of linearity (0.5–1000 ng/mL). Additionally, our data are more suitable than those reported by Drost et al. (1984) using similar cartridges. The superiority of our procedure can be justified by the higher capacity of the selected cartridge (Bond Elut C18 Certify®), which guaranteed higher recoveries of the drug during extraction from plasma samples.
Because derivatization is required prior to GC-MS analysis, a series of agents was proposed previously, such as bis-(trimethylsilyl)-trifluoroacetamide/BSTFA, trifluoroacetyl anhydride (TFA),19 pentafluoropropionic anhydride (PFPA)13,18, heptafluorobutirate anhydride (HFBA)15 and methoxamine plus propionic anhydride.31 The agent BSTFA was chosen in the present study and provided stable fragments, permitting GC-MS analysis for up to 8 hours after derivatization. The method validated in the present study showed results similar to data reported previously despite using different derivative agents. The higher sensitivity achieved in our study can be explained by the use of a fused silica capillary column that increases the sensitivity of the GC-MS method to analyze morphine, as discussed in previous studies.15,18,31–33
Thawing cycles in the present study showed good stability for morphine in plasma samples, and they are in agreement with the data described by Leis et al., 2000. Finally, none of the procedures reported previously described the short term stability of morphine or its internal standard solutions in methanol and also in plasma samples. The robustness of the analytical method investigated in this study has no available previously reported data for comparison.
Finally, the present method is possibly more costly than most of the current analytical chromatographic assays, but, as is the case of most instrumental methods, the cost is significantly reduced as the number of analyses scales up. In addition, the intrinsic advantages of the present method certainly imply a gain in terms of the cost/benefit relation.
Application of the GC-MS method to monitor morphine plasma levels in post-surgical patientsAfter total morphine PCA delivered doses of 19.5 mg, plasma concentrations of morphine obtained by GC-MS in post-surgical patients expressed as medians were: 13.1 ng/ mL (3rd h), 18.9 ng/mL (6th h), 53.0 ng/mL (12th h), 28.9 ng/ mL (18th h), 13.9 ng/mL (24th h) and 15.4 ng/mL (36th h). The peak morphine plasma concentration was reached at the 12th hour and declined up to the 24th hour. Drug consumption in each period indicated that morphine consumption increases in the late postoperative period, between 24 and 36 hours.
In a previous clinical study, Dal et al. (2003) compared two groups of surgical patients with a morphine PCA device pump for analgesia purposes in the postoperative period after a coronary artery bypass graft. The first group of patients received morphine PCA, and a second group received a combination of morphine PCA plus a basal drug infusion. Drug plasma concentrations did not show a difference between groups: for group 1 and group 2, concentrations were 80 and 86 ng/mL (4th h), 86 and 119 ng/mL (20th h), 115 and 153 ng/mL (28th h), 104 and 125 ng/mL (44th h), respectively. Additionally, the degree of analgesia between groups was not different, but the total morphine consumption between groups differed from the 28th h up to 44th h.37
In the present study, our data show that morphine plasma concentrations during the postoperative period of cardiac surgery were lower than those reported by Dal et al 2003. In fact, these differences could be described as a consequence of the higher selectivity of our analytical procedure (GC-MS) compared to the method reported previously, which was based on chemoluminescence measurements that determine the concentration of morphine plus its main metabolite, morphine-6-glucuronide.37
CONCLUSIONThe advantages of the method described in the present study include the selective determination of morphine using GC-MS, a reduced plasma volume requirement, high sensitivity due to optimal recovery of drug by solid phase extraction and robust chromatographic assay. In addition, the short run-time permits its application in clinical studies, including drug plasma monitoring, optimisation of PCA device for intermittent boluses delivery of morphine for short or long term periods and also for pharmacokinetic studies.
The project was supported by the Coordination for the Improvement of Higher Education Personnel (CAPES, Brazil).