



To investigate a possible direct, growth hormone-releasing, hormone-independent action of a growth hormone secretagogue, GHRP-2, in pituitary somatotroph cells in the presence of inactive growth hormone-releasing hormone receptors.
MATERIALS AND METHODS:The responses of serum growth hormone to acutely injected growth hormone-releasing P-2 in lit/lit mice, which represent a model of GH deficiency arising from mutated growth hormone-releasing hormone-receptors, were compared to those observed in the heterozygous (lit/+) littermates and wild-type (+/+) C57BL/6J mice.
RESULTS:After the administration of 10 mcg of growth hormone-releasing P-2 to lit/lit mice, a growth hormone release of 9.3±1.5 ng/ml was observed compared with 1.04±1.15 ng/ml in controls (p<0.001). In comparison, an intermediate growth hormone release of 34.5±9.7 ng/ml and a higher growth hormone release of 163±46 ng/ml were induced in the lit/+ mice and wild-type mice, respectively. Thus, GHRP-2 stimulated growth hormone in the lit/lit mice, and the release of growth hormone in vivo may be only partially dependent on growth hormone-releasing hormone. Additionally, the plasma leptin and ghrelin levels were evaluated in the lit/lit mice under basal and stimulated conditions.
CONCLUSIONS:Here, we have demonstrated that lit/lit mice, which harbor a germline mutation in the Growth hormone-releasing hormone gene, maintain a limited but statistically significant growth hormone elevation after exogenous stimulation with GHRP-2. The present data probably reflect a direct, growth hormone-independent effect on Growth hormone S (ghrelin) stimulation in the remaining pituitary somatotrophs of little mice that is mediated by growth hormone S-R 1a.
The synthesis and secretion of growth hormone (GH) are primarily regulated by the hypothalamic hormones GH-releasing hormone (GHRH) and somatostatin by the negative feedback of GH and IGF-I and by the natural endogenous GH-releasing hormone ghrelin (1–8). Normal somatotroph maturation, proliferation, and somatic growth and development require GHRH (9). In the late differentiation phases of somatotroph cells, GHRH activates Gs alpha, cAMP, and the protein kinase A pathway through its cell membrane receptor GHRH-R (1,10,11). Conversely, ghrelin, which was initially isolated from the rat stomach and hypothalamus, acts through the growth hormone secretagogue (GHS) receptor (GHS-R 1a), which is coupled to members of the Gq/i family and activates phospholipase C (2,12,13).
Synthetic GHSs are ghrelin receptor agonists that stimulate GH secretion in vitro and in vivo. They include GH-releasing peptides (GHRPs), such as GHRP-2, and non-peptide compounds. Synthetic GHSs and ghrelin also stimulate ACTH/cortisol and prolactin release via hypothalamic effects and have been shown to increase food intake, energy expenditure, sleep and cardiac tone (15–17). Although their chemical structures vary, all GHSs seem to act through the GHS-R to enhance GH secretion and food intake. GHS-R mRNA has been identified in the pituitary gland, arcuate nucleus of the hypothalamus, and in other tissues (6),. For maximal GH stimulation, GHRPs require a simultaneous secretion of hypothalamic GHRH (18–21). Furthermore, ghrelin and synthetic GHSs potentiate GHRH-induced cAMP production and increase the levels of several GHRH-Rs, which may also result in altered interactions between GHS-R and GHRH (22–25). In this context, a dual and complementary action of ghrelin and GHRP on the hypothalamus and pituitary gland may occur. Furthermore, a paracrine effect on the arcuate nucleus of the hypothalamus, which is involved in food intake and energy expenditure regulation, should be mentioned (17).
The predominant site of action of GHSs for GH secretion has not been fully established. GHS-Rs have been identified in both the pituitary gland and the hypothalamus, and GHSs may act at either or both sites. Some evidence indicates that the GH-secreting action of GHSs and ghrelin may occur mainly in the hypothalamus, with a marginal direct effect on the pituitary gland (25–27). Accordingly, a limited but statistically significant GH elevation (p<0.05) was first documented following an acute stimulus with GHRP2 in GH-deficient patients with short stature resulting from a germline mutation in the GHRH-R gene (28). Soon after, other studies supported these initial findings (29,30), which indicates a direct action of GHRP-2 in the pituitary gland. The cloning of the mouse and human growth hormone-releasing hormone receptor gene (ghrhr) was accomplished following the finding that different mutations analogous to the ghrhr Asp60Gly inactivating mutation of GH-deficient little mice also occurred in GH-deficient humans (31–37). The ghrhr mutation in lit/lit mice resulted in a complete lack of GHRH-mediated GH release, which was further supported by the finding that the mutated GHRH-R protein did not bind to GHRH (32,33). As expected, the complete lack of a GH response to GHRH1-29NH2 has been reported in little mice (38–40). The complete lack of a GH response to GHRP-6 was also reported in little mice (41). Nevertheless, because GHSs act specifically through the GHS-R, it is reasonable to assume that a GH response may occur in response to a GHS challenge in little mice. In support of this hypothesis, GHRP-2 has been shown to increase the GH release from rat pituitaries in vitro, even after the GHRH-R was blocked using the antisense oligonucleotide approach (42).
The little mouse is an established animal model for assessing the direct GHRH-independent effects of GHSs and ghrelin on pituitary somatotroph cells (33–35). Here we evaluated the GH response to GHRP-2 in little mice (lit/lit), their heterozygous (lit/+) littermates, and wild-type (+/+) controls. Additionally, a possible role of GHRH and GHRH-R on the secretion and action of ghrelin was evaluated by measuring the fasting and fed plasma ghrelin and serum leptin levels in these animals.
MATERIALS AND METHODSAnimals and housing conditionsLittle mice (C57BL/6J lit/lit) and their heterozygous (lit/+) littermates were purchased from The Jackson Laboratory (Bar Harbor, ME, USA), and a breeding colony was established in our animal house (43). Mutant mice were produced by mating C57BL/6J lit/lit females to C57BL/6J lit/+ males. Mice of both sexes at 45-90 days of age were used in the assays. As controls, wild-type (+/+) C57BL mice, obtained from the Tropical Medicine Division, University of São Paulo School of Medicine (São Paulo, Brazil), were used at 45-90 days of age (body weight ∼30 g). The body weights of the lit/lit and lit/+ mice were approximately 10-12 g and 20-25 g, respectively, at the same age and were obtained by a sensitive method (43).
The mice were maintained in an air-conditioned room at a temperature of 24±1°C. Water and food were provided ad libitum, and light was regulated on a 12-h light/12-h dark schedule.
A group of mice was kept under fasting conditions by removing food in the afternoon at approximately 17:00 h. Blood was collected from the fasted and non-fasted mice groups the following day at 09:00 h. Water was provided ad libitum to the fasted mice (43).
This research was approved by the local ethics committee and met the criteria for animal experiments.
Sequencing analysisGenotyping was performed to confirm and genetically characterize the lit/lit, lit/+, and wt/wt mice. The mice were genotyped by the PCR amplification of tail DNA isolated using the standard phenol protocol of our laboratory (44,45). Two primers, 5′-TGAGCTTGCATGTCTTCAGG-3′ and 5′-GGGATTAGACCAGCCAGTGA-3′ (annealing temperature, 60°C), were used to amplify the gene region of the ghrhr Asp60Gly mutation that results in the little mouse phenotype (24). The mutation analysis was performed by automated sequencing using the Big Dye Terminator v3.1 (310 Sequencer, Applied Biosystems, Foster City, CA, USA).
The forward and reverse strands were analyzed in duplicate DNA samples. Two sequencing editor software programs (Gene StudioTM Professional Edition, Suwanee; and Mutation Surveyour, Softgenetics, PA, USA) were used to identify DNA abnormalities.
Preparations and injectionsGHRP-2 (200 µg/ml; lot KP102-HQ007; Pramorelin: D-alanyl-3-(2-naphthyl)-D-alanyl-L-alanyl-L-tryptophyl-D-phenylalanyl-L-lysinamide dihydrochloride) was obtained from Fujisawa USA, Inc. (Deerfield, IL). The mice were given intra-peritoneal (i.p.) injections and weighed. Depending on the experimental design, blood samples were collected from the orbital sinus for the measurements of GH, IGF-I, leptin, or ghrelin.
ImmunoassaysThe mouse GH (mGH) serum levels were determined by a highly sensitive in-house specific RIA using reagents obtained from Dr. A. F. Parlow of the National Hormone and Pituitary Program (NHPP, Torrance, CA) (46). All samples were assayed in duplicate. In each assay, pools of wt/wt C57BL and lit/lit mice sera were used as internal controls. Eight mice from each strain were used, and the pooled (n = 8) serum level of GH was 1.3±0.7 ng/ml in the lit/lit mice and 6.5±1.8 ng/ml in the wt/wt C57BL mice. The initial GH levels in the lit/+ mice were not significantly different from those in the wt/wt C57BL mice, probably because of the number of animals. The sensitivity of the GH assay, which was performed via a delayed tracer addition, was 0.25±0.15 ng/ml as calculated according to Rodbard's definition (47). The inter-assay coefficient of variation was less than 10%.
Levels of IGF-I were determined in duplicate using a rat IGF-I RIA kit (DSL-2900, Diagnostic Systems Laboratories Inc., Webster, Texas, USA) with a minimum detection level of 21 ng/ml and measurable concentrations ranging from 150 to 4,500 ng/ml.
Leptin levels were determined using a rat leptin RIA kit (Linco Research Inc., St. Charles, MO, USA) with a minimum detection value of 0.2 ng/ml and a measurable concentration range of 0.2-12.8 ng/ml.
Total ghrelin (octanoylated + desoctanoylated)(48) levels were determined using a rat ghrelin RIA kit (Phoenix Pharmaceuticals Inc., Belmont, CA, USA) with a sensitivity of ∼5.4 pg/tube and a range of 1-128 pg/tube. Active ghrelin (octanoylated) levels were determined using the Linco Research kit (St. Charles, MO). For these measurements, blood samples were collected in tubes containing 0.78 mg K2EDTA (250-500 ml blood/tube), kept on ice and spun in a refrigerated centrifuge. PMSF (0.1 mg/ml plasma) and 1 M HCl (50 ml/ml plasma) were added to the plasma samples. The samples were kept frozen at -70°C until the ghrelin levels were measured. For the active ghrelin kit, the intra- and inter-assay coefficients of variation were 6.5-9.5% and 9.6-16.2%, respectively. For the total ghrelin kit, the intra- and inter-assay coefficients of variation were <10% and <15%, respectively.
Study designTwo groups of age-matched, wild-type (+/+) C57BL mice were initially injected i.p. with either 1 µg (32 animals/group) or 10 µg (18 animals/group) GHRP-2, and blood samples for the GH measurements were collected at different times up to 1 h after injection. Three mice were used for each time point, and the blood was withdrawn only once before the animals were sacrificed. In a pilot study, 10 µg GHRP-2 was administered i.p. to 2-3 lit/lit, lit/+ and (wt/wt) mice (n = 18 animals/group), and the GH response was measured at different times up to 1 hour after injection to assess the effects of dose and timing.
An independent two-week study was performed with lit/lit mice treated daily with 10 µg GHRP-2 i.p. per mouse (n = 6 mice for the control and n = 8 mice for the experimental group). The body weight was measured, and blood was drawn every 3-4 days to measure the GH, IGF-I and leptin levels. As previously described, the body weight was measured using a precise, highly sensitive methodology (40). Briefly, animal body weights were measured daily throughout the entire experiment and then used to calculate individual growth curves; the slopes of the growth curves were then used as response parameters. For the lit/lit mice, the acceptable daily weight variation in the last ten days before each assay (pre-assay period) was within 0.0025±0.0045 g/day (36,40). Thus, approximately 10% of the homozygous dwarf mouse population was rejected. Our criteria for selecting the lit/lit mice were based on the growth curves of the homozygous (lit/lit) and heterozygous (lit/+) mice of the C57BL/6J strain, which had already been established in a previous study (36) and further confirmed by the genotyping performed here.
To evaluate and compare the areas under the curve of the hormonal values, an arbitrary unit of concentration (ng/ml/time (min)) was defined.
StatisticsTo analyze the leptin levels and hormonal responses as measured by the area under the curve (AUC) data, a two-way analysis of variance (ANOVA) was performed with time and treatment as the independent variables. Student's t-test with Bonferroni's adjustment was used for all values obtained after initiation of treatment. In all other measurements, Student's paired t-test was used to evaluate the significance of differences. p<0.05 was considered to be significant.
RESULTSghrhr mutation analysisDNA was extracted from a blood sample of a lit/lit mouse using routine methods described elsewhere (44,45). PCR using previously reported primers was performed, followed by the sequence analysis of the ghrhr gene.
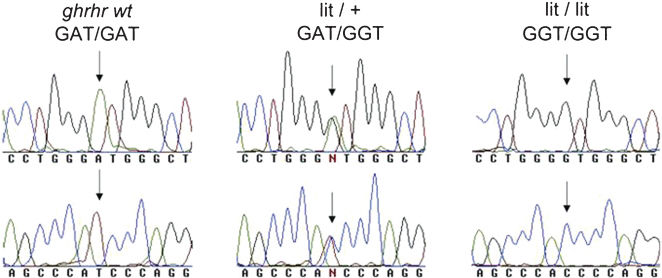
A germline homozygous ghrhr Asp60Gly mutation was confirmed in the lit/lit mouse, whereas a heterozygous ghrhr Asp60Gly mutation was verified in the lit/+ littermates. Conversely, wild-type (+/+) C57BL/6J mice did not harbor this germline mutation (Figure 1). These data confirmed the genotypes of the three mice lineages used in the present study, respectively: +/+, Asp60Asp; lit/+, Asp60Gly and lit/lit, Gly60Gly (Figure 1).
, GAT/GGT (heterozygous mouse) and GGT/GGT (little mouse) statuses, thus characterizing the genotype of the three mice strains used in this study. The forward and reverse traces are shown in the top and bottom panels, respectively.")
The germline ghrh-r Asp60Gly mutation analysis was performed by PCR amplification and direct automated sequencing. The chromatogram traces show the GAT/GAT (wild-type), GAT/GGT (heterozygous mouse) and GGT/GGT (little mouse) statuses, thus characterizing the genotype of the three mice strains used in this study. The forward and reverse traces are shown in the top and bottom panels, respectively.
The mean basal mGH levels in the 18 homozygous lit/lit mice (1.16±0.97 ng/ml; range, 0.28-4.0 ng/ml) were significantly lower than those observed in the 46 wild-type (+/+) controls (5.36±2.60 ng/ml; range, 1.05-14.0 ng/ml; p<0.001) or their 12 heterozygous (lit/+) littermates (6.60±2.35 ng/ml; range, 4.25-11.5 ng/ml; p<0.001). Furthermore, the basal GH concentrations observed in the lit/+ and +/+ mice did not differ significantly.
The basal serum IGF-I value in the 8 lit/lit mice (231±103 ng/ml; range, 120-420) was clearly lower than that in the 8 +/+ controls (473±104 ng/ml; range, 330-560 ng/ml; p<0.001).
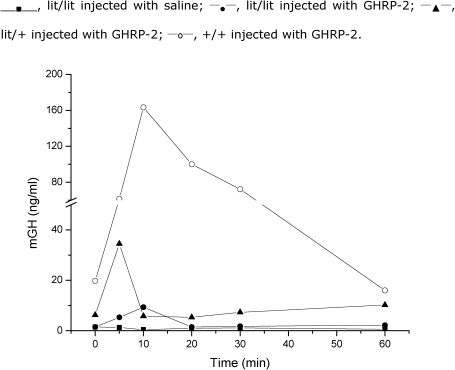
Response of GH to acute GHRP-2 administrationIt was initially shown that the increase of GH in the C57BL wild type control mice in response to 1 µg GHRP-2 was significantly less (∼two-fold increase over basal levels) than the increase in response to 10 µg of i.p. GHRp-2 (24-fold increase over baseline values; p<0.05). Thus, the 10-µg GHRP-2 dose was selected for the assessment of the GH responses in the three groups of mice (+/+, lit/+ and lit/lit).
The mean peak GH response in the control group (+/+) was 163 ng/ml, and it occurred 10 min after the administration of GHRP-2 with a 24-fold increase over the baseline levels (Figure 2). In the lit/+ and lit/lit mice, there was a significant GH response to GHRP-2 at 5-10 min. Twenty minutes after GHRP-2 injection, GH levels returned to the baseline values.
 serum concentrations were determined by a sensitive RIA method (detection limit, 0.25 ng/ml). Shown here are the mGH increases after the acute administration of 10 µg GHRP-2 in lit/lit, lit/+ and +/+ mice as well as the saline injection of lit/lit mice. Three animals were used for each time point only once each and then sacrificed. In total, 18 animals were used for each of the four observations, and 72 animals were used in total.")
Mouse GH (mGH) serum concentrations were determined by a sensitive RIA method (detection limit, 0.25 ng/ml). Shown here are the mGH increases after the acute administration of 10 µg GHRP-2 in lit/lit, lit/+ and +/+ mice as well as the saline injection of lit/lit mice. Three animals were used for each time point only once each and then sacrificed. In total, 18 animals were used for each of the four observations, and 72 animals were used in total.
These data indicate that the GH response to 10 µg GHRP-2 (9.3±1.5 ng/ml; range 8-11 ng/ml) observed in the homozygous lit/lit mice was significantly higher than that in the saline-injected lit/lit mice (1.04±1.15 ng/ml; p<0.001). Furthermore, the response of GH to GHRP-2 in the lit/lit mice was significantly lower than that observed in the heterozygous (lit/+) littermates (9.3±1.5 ng/ml vs. 34.5±9.7 ng/ml; p<0.01) or in the wild type (+/+) mice (163±46 ng/ml; p<0.005). Thus, the GH response to GHRP-2 in the lit/+ mice was intermediate to those observed in the lit/lit and +/+ mice (Figure 2).
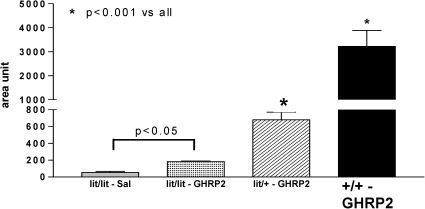
The absolute GH increases over the baseline in the lit/lit, lit/+, and +/+ mice were 8.3, 28.8, and 156 ng/ml, respectively, which confirms the marked differences in GH secretion among the three studied mice strains. The increase in the GH level over the baseline following the acute administration of 10 µg GHRP-2 did not differ significantly between the lit/lit and lit/+ mice (8.9- vs. 6.1-fold; p>0.05). However, the increase in GH in response to GHRP-2 was significantly decreased in the lit/lit (p<0.005) and lit/+ mice (p<0.01) compared with the controls. The individual areas under the curve (AUCs) of the serum GH in response to the acute administration of GHRP-2 further confirmed the significant differences between the GHRP-2- and saline-treated lit/lit mice (Figure 3).
 in the lit/lit, lit/+, and +/+ mice expressed as the areas under the curve (AUCs). An arbitrary unit (ng/ml/min) was used to allow evaluation of the AUC. The animal experiments are as shown in Figure 2.")
Serum mGH responses to the acute administration of 10 µg GHRP-2 (or saline, SAL for lit/lit mice) in the lit/lit, lit/+, and +/+ mice expressed as the areas under the curve (AUCs). An arbitrary unit (ng/ml/min) was used to allow evaluation of the AUC. The animal experiments are as shown in Figure 2.
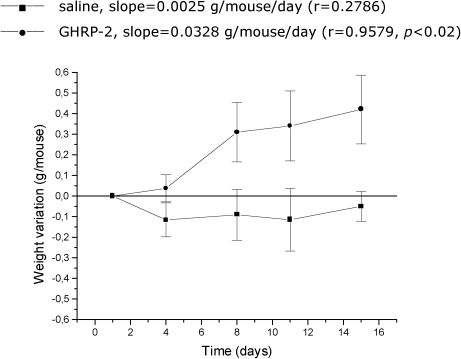
As expected, a marked increase in body weight was observed in the lit/lit mice when 10 µg GHRP-2 was i.p. injected (49) once a day for two weeks in comparison to injection of the control vehicle (p<0.02; Figure 4). However, GH and IGF-I levels did not significantly increase during the two-week administration of GHRP-2 (data not shown). Furthermore, markedly elevated (p<0.005) leptin levels in the lit/lit mice were observed on day 15 of the GHRP-2 treatment.
. The control group was injected with saline; six lit/lit mice were used in this experiment (n = 6). The mean and standard error of the mean intervals are shown.")
The weight gain assay upon the chronic administration of 10 µg/animal/day GHRP-2 in the lit/lit mice; eight animals were used in this experiment (n = 8). The control group was injected with saline; six lit/lit mice were used in this experiment (n = 6). The mean and standard error of the mean intervals are shown.
The concentrations of plasma acyl ghrelin and total (acyl + deacyl) ghrelin, which may have independent and interrelated biological actions, and the concentrations of serum leptin in the +/+, lit/+, and lit/lit mice in the fasted and fed state are shown in Table 1. In the fasted state, the mean plasma acyl ghrelin and total ghrelin levels in the lit/lit mice were significantly lower (by 78.9% and 37.6%, respectively) than in the +/+ mice. The heterozygous lit/+ mice also had significantly lower levels of fasting acyl ghrelin and total ghrelin (by 80.5% and 44.9%, respectively) compared with the controls. In the lit/lit mice, the plasma acyl ghrelin in the fed state was 93.8% higher than in the fasted state. Conversely, in the wild-type mice, the plasma acyl ghrelin was 82.5% lower (p<0.001), and total ghrelin was 61% lower (p<0.001) in the fed state than in the fasted state. The difference in the fed-state plasma acyl ghrelin levels between the two groups (157±70 pg/ml for +/+ mice vs. 67±18 pg/ml for lit/lit) was significant (p<0.05). Furthermore, the total ghrelin levels were 23.4% higher in the fasted state than in the fed state for the lit/lit mice.
The plasma ghrelin and serum leptin concentrations in fed and 16-hour-fasted little (lit/lit), heterozygous (lit/+), and wild-type (+/+) mice.
| Mouse strain | Bioactive ghrelin (pg/ml) | Total ghrelin (pg/ml) | Leptin (ng/ml) | |||
|---|---|---|---|---|---|---|
| fed | fasted | fed | fasted | fed | fasted | |
| +/+ | 67 ± 18.2(6) | 384 ± 100 | 1272 ± 410(7) | 3261 ± 222 | 2.70 ± 0.32(8) | 2.32 ± 0.60 |
| lit/+ | 85 ± 14.2(NS) | 75 ± 3.0(1) | 1538 ± 96.0(NS) | 1796 ± 540(2) | 7.09 ± 5.0(9) | 5.18 ± 0.72(2)(5) |
| lit/lit | 157 ± 70(4) (10) | 81 ± 21.8(2) | 2513 ± 384(11) | 2036 ± 706(3) | 6.78 ± 3.74(12) | 10.40 ± 2.62(2) |
All measurements are derived from the average ± SD of n = 4 animals/group.
(1) p<0.001, (2) p<0.005, (3) p<0.02, (4) p<0.05, NS = non-significant, all compared to +/+; (5) p<0.02 compared to lit/lit; (6) p<0.001 compared to fasted; (7) p<0.001 compared to fasted; (8)-(12) all NS compared to the corresponding fasted animals.
The serum leptin levels of the fasted lit/lit mice were 348% higher than those in the fasted +/+ mice (p<0.005). The fasted lit/+ mice also differed from the lit/lit (50% lower, p<0.02) and +/+ mice (123% higher, p<0.005). Moreover, in the lit/lit mice, the serum leptin levels in the fed state decreased by 35% compared with the fasted state (p>0.05). Additionally, leptin levels in the fed lit/+ and +/+ fed mice were 37% and 16% higher than in the fasted animals, respectively (p>0.05).
DISCUSSIONSeveral spontaneous homozygous germline mutations in mice that result in the deficiency of pituitary hormones and dwarfism have been documented (34). Thus, the phenotypes of the Ames dwarf mice result from mutations in the Prop1 gene and present a congenital deficiency of multiple pituitary hormones, including GH (50). In addition, Snell dwarf mice with mutations in the pit gene have dwarfism arising from GH deficiency, hypothyroidism, and infertility (51). Furthermore, the little mice phenotype results from homozygous mutations in the ghrh-r gene (33). Equivalent germline homozygous mutations in the PROP1, PIT, and GHRH-R genes have been reported in humans presenting with severe short stature (37,52,53).
GHRH-R mutations have been specifically suggested to cause the absence of a GH response to GHRP-2 in humans (54). However, several other studies documented a limited but statistically significant increase in GH after the administration of GHRP-2 to these patients (28–30). Similarly, it has been suggested that the little mouse is resistant to the action of GHRP-2 and does not present an increase in GH after the administration of this peptide (41). Thus, the main goal of the present study was to investigate whether little mice have an absolute resistance to the action of GHRPs, as previously described, or if they present a statistically significant GH response to one of the GHRPs, i.e., GHRP-2, as observed in humans. To detect minor GH variations in the serum, we used an improved mGH method to better discriminate possible GH increases after the administration of GHRP-2.
To our knowledge, no other paper to date has specifically re-investigated this issue in little mice. Of note, complete GH resistance to GHRP-2 in ghrh-knockout mice has been documented. However, the ghrh-knockout mice and little mice have mutations in different genes, although their phenotypes are similar. Furthermore, different GH methods and experimental conditions may have influenced these apparently contradictory results.
GH responses to GHRP-2The development and function of somatotroph cells are GHRH-dependent (1), as indicated by our findings of a limited GH response to acute GHRP-2 administration in lit/lit mice carrying a homozygous mutation in ghrhr. Assuming that ghrhr is completely inactive in lit/lit mice (39), our present findings indicate that at least some GHRH-independent GHS-GH release occurs through the activation of the GHS-R.
In addition to mature GHRH-dependent somatotrophs, GH-producing stem cells have been found in the pituitary glands of lit/lit mice and in 60-day-old adult mice (26). However, it is not known whether GHRP-2 and/or ghrelin act as a trophic factor for the GH-producing stem cells independently of the pituitary action of GHRH.
The previously reported lack of a GH response in lit/lit mice to another type of GHS, GHRP-6 (41), may be related to the use of a less sensitive GH assay (10 ng/ml vs. 0.25 ng/ml for our assay). Furthermore, GHRP-2 has a greater biological potency (approximately six-fold greater) than GHRP-6 for triggering the release GH (14–16). The absence of GH responses to GHRP-6 was also initially suggested in humans with a Glu72Stop GHRH-R mutation (42). However, several other studies have documented the presence of a statistically significant, although impaired, GH response to GHRP-2 in patients with genetic short stature who harbor a severely truncated GHRHR gene (28–30).
Furthermore, neither acute nor chronic GH elevations were noticed in ghrh-knockout mice, and it was concluded that GHRP-2 has a growth-stimulating effect that augments the response induced by JI-38 (55,56). However, it is important to note that a) knockout mice and animals harboring similar or even equivalent spontaneous germline mutations may behave differently; b) the mGH methods used to study the little mice and knockout mice could be different; and c) different experimental designs were used in these studies (28–30,55,56).
The intermediate GH responses to GHRP-2 in the lit/+ mice may be the result of qualitative and/or quantitative differences in the somatotroph cells, although further research on this topic should be conducted to confirm these findings. These data may suggest a genetic dosage effect on somatotroph cell function, which would become more impaired as a function of age. Similarly, a genetic dosage effect was previously proposed for cases carrying a mutation in the GHRHR gene (52).
Importantly, our GH RIA method was able to detect the presence of very low GH levels (∼0.25 ng/ml) in little mice with acceptable precision; similar data are seldom available in the literature. GH kits with lower sensitivities have been used by others. However, no other reports were found that addressed the serum GH levels of lit/lit mice obtained by a specific homologous RIA. Cheng et al. reported serum GH levels of 0.61±0.09 ng/ml in male and female lit/lit mice and 8.50±0.75 ng/ml and 2.85±0.33 ng/ml in male and female lit/+ mice, respectively. Marmary et al. reported serum GH levels of 1.08±0.06 ng/ml and 20.35±22.9 ng/ml in Snell dwarf mice and their control littermates, respectively (58). High estimates of the absolute level of serum GH determined using a heterologous rat GH RIA have also been reported (59,60).
Growth following GHRP-2 administrationIn this study, a definite increase in the body weight of little mice was observed after the administration of GHRP-2. These findings are in agreement with other studies that documented a significant body weight gain in ghrh-knockout mice treated with GHRP-2 and in mice after the administration of ghrelin, which shows that both peptides induce adiposity in these animals (61). Of note, Alba et al. did not observe an increase in the longitudinal growth of ghrh-knockout mice during a six-week treatment with GHRP-2 at a dose of 10 µg injected subcutaneously (s.c.) twice a day, which confirmed the pivotal role of GHRH in GH secretion and subsequent body growth (48). Additionally, a chronic six-week treatment with 10 µg s.c. GHRP-2 did not increase the acute GH response (30-min). This apparent discrepancy with our findings may have resulted from differences in the time of blood collection or GH assay sensitivity, different methods of drug administration (s.c. vs. i.p.), or a combination of these factors.
Leptin and ghrelin levels in the lit/lit miceWe also evaluated whether GHRP-2 would influence leptin and ghrelin levels.
The fasting levels of acyl ghrelin and total ghrelin were higher in the wild-type +/+ mice than in the fed mice (61–63). The physiological regulation of the plasma ghrelin (i.e., its increase and decrease in the fasting and fed states, respectively) was different in the lit/lit mice. This finding may be the result of a lack of effect of the action of GHRH and its inactive GHRH-R, although this hypothesis should be tested further.
High basal serum leptin levels and an additional increase after prolonged GHRP-2 administration in lit/lit mice have also been observed in mice treated with ipamorelin (34). These findings are considered to reflect the increased adipose tissue mass in GH-deficient mice and the adipogenic effect of GHRP-2 and ghrelin (61–63).
To investigate the lipogenic effects of these peptides in mice, the doses of GHRP-2 used in the present study were similar to those used by Tschop et al. and to the GHRP-6 doses used by Jansson et al. and Lall et al. (38,49,61). However, based on body weight, such doses were proportionally much higher than those tested in humans with a GHRH-R mutation, which could reflect interspecies differences in the sensitivity to GHSs (61).
Our limited data on the octanoyl ghrelin (ghrelin) and serum leptin levels in fed vs. fasted (17 h) wild-type (+/+) and lit/lit mice (-/-) may suggest dysfunctional relationships between these two metabolic hormones. As expected, the plasma ghrelin levels were normal in the fed wild-type mice and elevated in the fasted wild-type mice, although the opposite effect was also observed, as ghrelin and leptin were elevated in the fed state and did not further increase in the fasted state. Additionally, the results in the lit (+/-) mice partially paralleled those obtained in the lit (-/-) mice in that the plasma ghrelin levels did not increase with fasting, and during fasting, the serum leptin levels remained elevated in the fed and fasted lit (+/- and -/-) mice. However, the limited number of studied animals prevented further conclusions from being drawn. Finally, the measurement of total ghrelin as an indicator of octanoyl ghrelin levels may be problematic. Although the total and octanoylated ghrelin levels may partialy parallel each other, this agreement becomes less evident under pathophysiological conditions, such as in the present mouse study. Increasing evidence supports the biological activity of the desoctanoyl ghrelin molecule and thus in turn supports the measurement of the plasma desoctanoylated ghrelin levels by a specific assay, such as that published by Akamizu et al. (64).
In conclusion, our findings are the first to document the presence of statistically significant increases in GH following the administration of GHRP-2 in little mice. The data give further support to a direct action of GHRP-2 in the pituitary glands of little mice. Furthermore, the heterozygous lit/+ mice may have subtle disturbances in their GHRH/GHRH-R/GH axis, which suggests a genetic dosage effect, although additional data are needed to confirm this conclusion.
AUTHOR CONTRIBUTIONSPeroni CN, Nascimento N, and Bartolini P performed the experiments and collaborated in the manuscript preparation. Hayashida CY collaborated in the manuscript writing. Toledo RA and Longuini VC performed the genetic analyses. Bowers CY collaborated with the product testing (ghrp-2), the measurements of ghrelin and leptin and the preparation of the manuscript. Toledo SP wrote and coordinated the research project and was the senior researcher responsible for preparing the manuscript.
We acknowledge Cristina T. Kanamura, B.S., and Venâncio A. F. Alves, M.D., from the Division of Pathology, Institute Adolfo Lutz, São Paulo, for technical support. We would like to thank Dr. Heitor F. Andrade Jr., who provided the C57BL wt/wt animals. This research was partially supported by FAPESP (01/11091-0). S.P.A.T. and P.B. are National Research Council (CNPq) researchers. RAT is recipient of a FAPESP post-doctoral fellowship (2009/15386-6). SPAT is recipient of a CNPq fellowship and grant (401990/2010-9).
No potential conflict of interest was reported.