




Limb-girdle muscular dystrophy presents with heterogeneous clinical and molecular features. The primary characteristic of this disorder is proximal muscular weakness with variable age of onset, speed of progression, and intensity of symptoms. Sarcoglycanopathies, which are a subgroup of the limb-girdle muscular dystrophies, are caused by mutations in sarcoglycan genes. Mutations in these genes cause secondary deficiencies in other proteins, due to the instability of the dystrophin-glycoprotein complex. Therefore, determining the etiology of a given sarcoglycanopathy requires costly and occasionally inaccessible molecular methods.
OBJECTIVE:The aim of this study was to identify phenotypic differences among limb-girdle muscular dystrophy patients who were grouped according to the immunohistochemical phenotypes for the four sarcoglycans.
METHODS:To identify phenotypic differences among patients with different types of sarcoglycanopathies, a questionnaire was used and the muscle strength and range of motion of nine joints in 45 patients recruited from the Department of Neurology – HC-FMUSP (Clinics Hospital of the Faculty of Medicine of the University of São Paulo) were evaluated. The findings obtained from these analyses were compared with the results of the immunohistochemical findings.
RESULTS:The patients were divided into the following groups based on the immunohistochemical findings: α-sarcoglycanopathies (16 patients), β-sarcoglycanopathies (1 patient), γ-sarcoglycanopathies (5 patients), and non-sarcoglycanopathies (23 patients). The muscle strength analysis revealed significant differences for both upper and lower limb muscles, particularly the shoulder and hip muscles, as expected. No pattern of joint contractures was found among the four groups analyzed, even within the same family. However, a high frequency of tiptoe gait was observed in patients with α-sarcoglycanopathies, while calf pseudo-hypertrophy was most common in patients with non-sarcoglycanopathies. The α-sarcoglycanopathy patients presented with more severe muscle weakness than did γ-sarcoglycanopathy patients.
CONCLUSION:The clinical differences observed in this study, which were associated with the immunohistochemical findings, may help to prioritize the mutational investigation of sarcoglycan genes.
Muscular dystrophy is a necrotic degenerative/regenerative process of the muscles and results in progressive muscle weakness and wasting. The mechanisms by which various molecular defects result in muscular dystrophy are not yet fully understood.1
Limb-girdle muscular dystrophies (LGMD) are clearly distinct from other muscular disorders, such as dystrophinopathies, myotonic disorders, or facioscapulohumeral dystrophies.2 The clinical course of LGMD is characterized by normal intelligence and great variability in muscle weakness and wasting, ranging from mild to severe forms. LGMD may show an early onset in the first decade of life with rapid disease progression or a later onset with slower disease progression.1,3 Hyperlordosis,4 scapular winging (escapula alata),5,6 tendon contractures,7 and a tiptoe gait pattern4 with a wide-based stance7 are associated with this disease. Serum creatine kinase (CK) levels are elevated in most cases, and patients may develop cardiomyopathies and/or respiratory insufficiencies.6
Due to the heterogeneity of LGMD and the lack of diagnostic specificity, estimates of the prevalence of all forms of LGMD range from 1/14,500 to 1/123,000.2 Autosomal dominant LGMD is relatively rare (5 loci described),8 but cases of autosomal recessive LGMD have been characterized worldwide and are a heterogeneous group of disorders that lead to progressive muscle wasting and weakness. Current evidence suggests the involvement of at least 14 distinct loci in autosomal recessive LGMD (http://www.musclegenetable.org).9
LGMD2C – F are autosomal recessive LGMDs, also known as sarcoglycanopathies (SGP), which are caused by mutations in the genes encoding the γ-, α-, β-, and δ-sarcoglycan proteins (SG), respectively.10–13 The SGs are glycosylated proteins with single transmembrane domains,14 and correct assembly of the sarcoglycan complex is required for the maintenance of the sarcolemma.15,16 Together with sarcospan, dystrophin, dystroglycans, syntrophins, and α-dystrobrevin, the SGs constitute the dystrophin-glycoprotein complex (DGC).17 The DGC acts as a link between the muscle cell cytoskeleton and the extracellular matrix, providing mechanical support for the plasma membrane during myofiber contraction.14,18 The function of the SGs is not fully understood, but they appear to play both mechanical and non-mechanical roles that mediate interactions among the extracellular matrix, the sarcolemma and the cytoskeleton.19,20 A primary mutation in any one of the sarcoglycan genes (α, β, γ or δ) can lead to the total or partial loss of that sarcoglycan, secondary deficiencies of the other sarcoglycans and the occasional reduction of dystrophin labeling in muscle tissue.21 Inter- and intra-familial heterogeneity is frequent.22 Mutations in the gene for α-sarcoglycan are the most common sarcoglycan mutations, whereas mutations in δ-sarcoglycan are the rarest.23
The phenotypes of the sarcoglycanopathies overlap with the dystrophinopathies with the important distinction that the learning disability specifically associated with Duchenne's is not present, and scapular winging is more frequent in SGPs.6,9,22,24
Some studies have shown distinct patterns of sarcoglycan expression and labeling in various SGPs,1,14,16,24 but we believe that further studies are required to obtain more conclusive information because the characteristics of each population vary greatly.
OBJECTIVESThe purpose of this study was to identify phenotypic differences among LGMD patients who were grouped according to the immunohistochemical findings for the four sarcoglycans.
MATERIALS AND METHODSPatientsForty-five patients (from 40 families) with clinical diagnoses of LGMD from the Department of Neurology – HC-FMUSP were studied. The clinical diagnosis was based on the presence of limb-girdle muscle weakness, muscle retractions, myopathic alterations determined by electromyography (EMG) and altered levels of serum creatine kinase (CK). In all of the patients included in this study, the diagnosis of dystrophinopathy was previously excluded by mutational analysis using the multiple polymerase chain reaction (PCR) method and protein analysis by immunohistochemistry and western blotting. This study was approved by the Institutional Review Board Ethics Committee on Human Experimentation of the Clinics Hospital of the University of São Paulo, which follows the guidelines of the Helsinki Declaration of 1975, and all subjects in this study signed an informed consent form.
Clinical EvaluationThe following clinical features were evaluated: age of onset, consanguinity, family history, maximum motor ability (ambulant, ambulant with aid, or confined to a wheel-chair), calf pseudo-hypertrophy, tiptoe gait pattern (Achilles tendon shortening), muscle strength, and the range of motion (ROM) of nine joints (to identify tendon contractures). The serum CK levels and the presence of cardiomyopathies, as determined by electrocardiography and echodoppler, were also evaluated.
Muscle strength was determined according to the Medical Research Council (MRC) scale, which ranges from 0 to 5. On this scale, grade 5 is considered normal strength; grade 4 signifies that the patient can complete resisted movements but does not have normal strength; grade 3 represents the ability to complete movements against gravity only; in grade 2 there is movement but not against gravity; grade 1 represents contraction without movement; and grade 0 denotes an absence of muscle contraction. The MRC scale was applied to evaluate the muscle groups involved in essential movements because all of the patients included in the present study were in a chronic stage of the disease, which posed difficulties in assessing individual muscle strength. The muscles involved in the following movements were evaluated: flexion and extension of the shoulders, elbows, wrists, fingers, thumbs, hips, knees, ankles, and toes; specific movements of each joint, such as shoulder and hip adduction, abduction, internal rotation and external rotation, shoulder elevation, forearm pronation and supination, wrist ulnar deviation and radial deviation, finger interosseous, thumb opponency, and ankle inversion and eversion.
To determine the presence of contractures, the patients were given a score of complete or incomplete ROM for each joint, and each individual joint and the percentage of compromised joints were considered. The joints that were evaluated for contractures were also evaluated for muscle strength.
Muscle BiopsyMuscle biopsies were obtained from the brachial biceps of all patients using local anesthesia in the operating room of HC-FMUSP. The samples were frozen in liquid nitrogen, and 6-μm thick sequential slices were collected on a cryostat. Routine histological and histochemical staining was conducted as described in detail by Dubowitz (1985).23
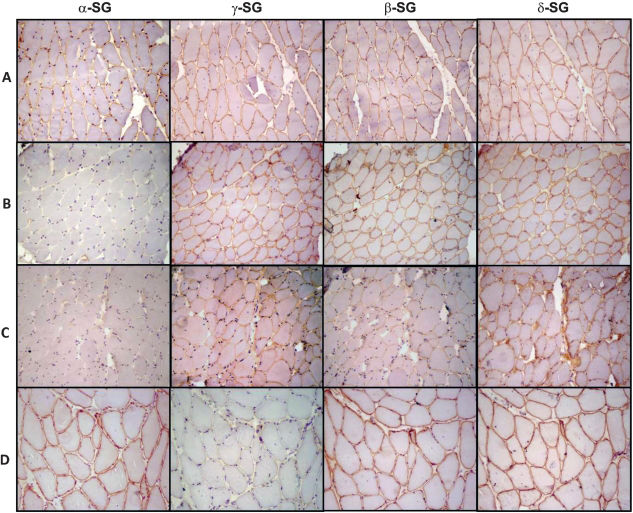
ImmunohistochemistrySpecific antibodies against the sarcoglycans were used to identify the pattern of staining of this protein complex. Primary antibodies against the following proteins were used: dystrophin carboxy terminus, amino terminus, and rod domain (Novocastra, Dy8/6C5, Dy10/12B2 and Dy4/6D3, respectively), diluted 1/1000; α-sarcoglycan (Novocastra, Ad1/20A6), diluted 1/100; β-sarcoglycan (Novocastra, bSarc/5B1), diluted 1/100; γ-sarcoglycan (Novocastra, 35DAG/21B5), diluted 1/100; and δ-sarcoglycan (Novocastra, dSarc3/12CI), diluted 1/100. The details of the immunohistochemical methods have been previously described by Ferreira et al. (2005).25 The sections were classified according to the intensity of the staining of the sarcolemma as follows: positive staining (complete staining of all fibers), patchy staining (partial or incomplete staining of most fibers) or negative staining (absence of staining of cell membrane). The samples with the lowest or absent immunoreactivity were classified as SG deficient, as shown in Figure 1. Examples of immunostained muscle biopsy samples from patients included in this series are presented in Figure 2.

. (A) A representative non-SGP case showing positive reactions for all four SGs; (B) a representative case of α-SGP showing no expression of α-SG and positive reactions for the remaining SGs; (C) a representative case classified as α-SGP showing patchy expression of all four SGs with lower expression of α-SG; and (D) a representative case of γ-SGP showing a negative reaction for γ-SG and positive staining for the other three SGs. 200x magnification. SG: sarcoglycan; SGP: sarcoglycanopathy.")
Immunohistochemical preparations for alpha, gamma, beta and delta sarcoglycans (α-, γ-, β- and δ-SG, respectively). (A) A representative non-SGP case showing positive reactions for all four SGs; (B) a representative case of α-SGP showing no expression of α-SG and positive reactions for the remaining SGs; (C) a representative case classified as α-SGP showing patchy expression of all four SGs with lower expression of α-SG; and (D) a representative case of γ-SGP showing a negative reaction for γ-SG and positive staining for the other three SGs. 200x magnification. SG: sarcoglycan; SGP: sarcoglycanopathy.
The data are expressed as the mean ± SEM. The Kruskal-Wallis test was used to identify differences among the groups in the non-parametric clinical data. Pearson's chi-square test was also employed to test for associations between components of the clinical data. A one-way analysis of variance (ANOVA) with Tukey's post hoc test (Unequal N HSD) was applied after using Tukey's test to record outliers and verify the differences, the power of these differences and the size effects on muscle strength.
RESULTSThe patients were divided into the following groups according to the immunohistochemical staining patterns: α-sarcoglycanopathy (α-SGP) (n = 16), γ-sarcoglycanopathy (γ-SGP) (n = 5) and non-SGP. The patients were classified as non-SGP when the staining for all four sarcoglycans was positive (n = 23). Only one patient was diagnosed as having a β-sarcoglycanopathy, and this patient was excluded so as not to bias the results. The immunohistochemical characteristics of each patient are shown in Figure 1, and the data collected from the clinical evaluations of the patients included in this study are shown in Table 1.
Demographic and clinical data collected from the patients.
| Groups | |||
|---|---|---|---|
| non-SGP | α-SGP | γ-SGP | |
| n | 23 | 16 | 5 |
| Predominant sex | M (54.2%) | M (62.5%) | F (60%) |
| Age (mean±SD) | 32.9 (±13.9) | 33.8 (±16) | 43.4 (±17.1) |
| Age of Onset (mean±SD) | 18.3 (±11.1) | 17.3 (±13) | 29.3 (±20.1) |
| Consanguinity | 25% | 37.5% | 80% |
| Family history | 58.3% | 56.3% | 60% |
| Cardiomyopathy | 26% | 25% | 40% |
| CK | nl – 50x | 2 – 36x | nl – 17x |
| Tiptoe gait pattern | 54.6% | 75% | 0% |
| Maximum motor ability | A (77.3%) | A (56.3%) | A (100%) |
| Calf pseudo-hypertrophy | 77.3% | 43.8% | 0% |
| % of joints with affected ROM | 43.1% (±13.5) | 55% (±31.3) | 37% (±23.5) |
non-SGP: positive staining for all sarcoglycans; α-SGP: α-sarcoglycanopathy; γ-SGP: γ-sarcoglycanopathy; n: number of patients; CK: range of fold increase in serum creatine kinase, A: ambulant; ROM: range of motion; nl: normal.
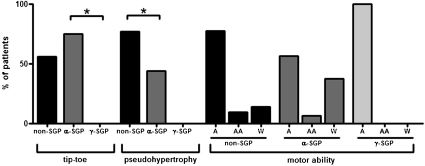
Differences in percentage of patients presenting with a tiptoe gait pattern and calf pseudo-hypertrophy were detected among the groups. The Kruskal-Wallis test revealed a significant difference in the percentage of patients presenting with a tiptoe gait pattern between the α-SGP and γ-SGP groups (p = 0.03) (Figure 3) and in the percentage of patients with calf pseudo-hypertrophy between the α-SGP and non-SGP groups (p = 0.02) (Figure 3). The maximum motor ability of the groups tended to vary. While 37.5% of the patients with an α-SG deficiency were confined to a wheelchair, only 12% of the patients with non-SGP and none of the γ-SG patients were wheelchair bound (Figure 3).
. Second, the percentage of patients in each group with calf pseudo-hypertrophy is shown. The Kruskal-Wallis test showed a significant difference between the α-SGP and γ-SGP groups (p = 0.02). Third, the maximum motor ability of the groups is shown. 37.5% of the patients with an α-SG deficiency were confined to a wheelchair, while only 12% of the patients with a γ-SG deficiency and none of the non-SGP patients were wheelchair bound. non-SGP: positive staining for all sarcoglycans; α-SGP: α-sarcoglycanopathy; γ-SGP: γ-sarcoglycanopathy; A: ambulant; AA: ambulant with aid; W: confined to a wheelchair.")
Histograms illustrating the clinical findings. First, the percentage of patients in each group presenting with a tiptoe gait pattern are shown. The Kruskal-Wallis test showed a significant difference between the α-SGP and γ-SGP groups (p = 0.03). Second, the percentage of patients in each group with calf pseudo-hypertrophy is shown. The Kruskal-Wallis test showed a significant difference between the α-SGP and γ-SGP groups (p = 0.02). Third, the maximum motor ability of the groups is shown. 37.5% of the patients with an α-SG deficiency were confined to a wheelchair, while only 12% of the patients with a γ-SG deficiency and none of the non-SGP patients were wheelchair bound. non-SGP: positive staining for all sarcoglycans; α-SGP: α-sarcoglycanopathy; γ-SGP: γ-sarcoglycanopathy; A: ambulant; AA: ambulant with aid; W: confined to a wheelchair.
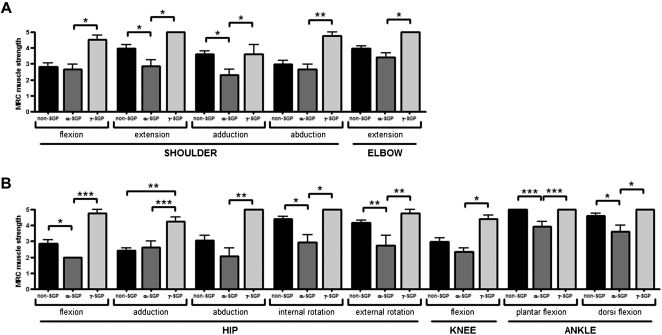
In general, the patients presented with symmetrical alterations in muscle strength and contractures with some exceptions due to falls or injuries affecting one side more than the other. Muscle weakness was more pronounced in the proximal than in the distal muscles as expected,7 and the flexor muscles of these patients were found to be more affected than the extensor muscles. The one-way ANOVA analysis for the comparison of muscle strength per segment showed significant differences among the groups (F(26.22) = 4.4; p = 0.0004). This analysis was highly significant (α = 0.99), and the partial eta squared (η2) value was equal to 0.84, indicating that 84% of the differences found were due to the diagnosis. The post hoc test (Tukey - Unequal N HSD) revealed some significant differences in the muscle strengths of the upper and lower limbs among the groups as depicted in Figure 4.
 and of the hips and lower limbs (B) (* p<0.05; ** p<0.01; *** p<0.001). non-SGP: positive staining for all sarcoglycans; α-SGP: α-sarcoglycanopathy; γ-SGP: γ-sarcoglycanopathy.")
Graphs illustrating the differences found among the three groups from the comparative analysis of the muscle strength scores of the shoulders and upper limbs (A) and of the hips and lower limbs (B) (* p<0.05; ** p<0.01; *** p<0.001). non-SGP: positive staining for all sarcoglycans; α-SGP: α-sarcoglycanopathy; γ-SGP: γ-sarcoglycanopathy.
The joints with decreased ROM, however, showed no characteristic contracture patterns among the three groups analyzed, even after comparing patients within the same family. Furthermore, no correlations between joint contractures and muscle strength were found.
DISCUSSIONThe present immunohistochemical study in a cohort of adult LGMD patients allowed us to classify 35% of cases as α-SGP, 12% as γ-SGP, 2% as β-SGP and 51% as non-SGP (in which no deficiency in any of the four SG proteins was found). All four SGPs appear to be prevalent in Brazil; however, δ-SGP is the least common form of autosomal recessive LGMD in Brazil26 and is equally rare worldwide.27,28 Accordingly, δ-SGP was not detected in the present cohort; the proportion of α-SGP and γ-SGP in our cohort was similar to the proportion found in a previous study.29
Phenotypic differences were identified among the α-SGP, γ-SGP and non-SGP groups. A tiptoe gait pattern was more frequent in the α-SGP group than in the other groups. The high frequency of this pattern may be due to the marked presence of contractures in the α-SGP subtype,7 including contractures of the Achilles tendon, which lead to the tiptoe gait pattern. Also, patients classified as α-SGP in the present cohort presented with more severe muscle weakness and were more frequently confined to a wheelchair than the other patients. In primary α-SGP, α-SG is the most severely reduced protein, although there are also deficiencies of the other three SGs.24,28–32 However, the spectrum of protein deficiency may vary from total absence to partial reduction of this protein with normal staining for the other SGs.32,33
In the present study, patients classified as γ-SGP had < mild phenotype with remarkably preserved muscle strength none of them was confined to a wheelchair or presented with a tiptoe gait pattern or calf pseudo-hypertrophy. These findings corroborate previous reports showing that γ-SGP has a mild phenotype despite the complete absence of γ-SG and a decrease in the other three SGs.21,28,33–35 However, in contrast to the present findings, some studies have reported that the early loss of ambulation, calf hypertrophy, contractures of the Achilles tendon, lumbar lordosis, scapular winging, and weak dorsal thigh and neck muscles are common clinical features of γ-SGP.36
Similar to α-SGP, studies on γ-SGP have described a heterogeneous phenotype. Nonetheless, the clinical variations from mild to severe disease have been correlated with the residual amount of γ-SG.33,37 Although the parameters governing the phenotype/genotype correlation remain unclear, a linear association has been described between the degree of protein deficiency and the onset of symptoms; a total absence of SGs is associated with an earlier mean age at disease onset than is observed with partial deficiencies.22
In the present cohort of patients, the patient presenting with a deficiency in all four SGs was classified as having α-SGP, as previously reported by others.24,28–32 However, this lack of all four SGs has also been described in a case confirmed to be γ-SGP by molecular testing.29 Nevertheless, the results and statistical analyses were not affected by the classification of this case as either α-SGP or γ-SGP.
The non-SGP group, which showed no immunohistochemical alterations in any of the four SGs, may include patients harboring either missense mutations in an SG gene (with no alterations in protein expression) or forms of LGMD other than SG deficiency. Therefore, patients with different etiologies were included in this group, which made the analysis difficult. However, the positive expression of all four SGs excludes the presence of null mutations in the SG genes in this group; this type of mutation leads to a more predictable, uniform and severe phenotype with decreased expression levels of the protein encoded by the affected gene.22 The higher frequency of calf pseudo-hypertrophy observed in this group, however, might point to some other form of LGMD, such as calpainopathy, in which calf pseudo-hypertrophy is common.7
Molecular testing is fundamental for the establishment of the final diagnosis of LGMD and will certainly be mandatory when a gene-based treatment becomes available. However, considering the continuous increase in LGMD types caused by alterations in different proteins of the muscular system, it is important to find clinical parameters and accessible laboratory tools to prioritize the molecular characterization of LGMD. Efforts have been made to improve the differential diagnosis by comparing mutations to immunohistochemical findings from SGP patients29 or by comparing general muscle strength to other laboratory data obtained from LGMD patients.38 Nonetheless, detailed muscular evaluations must be correlated with laboratory findings to identify clinical markers specific to each situation.
To this end, the present results show that a systematic clinical evaluation, together with immunohistochemical staining of muscle samples, enabled the characterization of LGMD patients. Applying this strategy to a larger number of patients may further refine these tools and help to determine the cost/benefit ratio of the molecular diagnosis.
This study was supported by CNPq (Brazil). The authors would like to thank Eliene Dutra Campos, Caroline Alencar and Thais Freire for their technical assistance.
No potential conflict of interest was reported.