



Extra-adrenal paragangliomas are very uncommon tumors arising from neuroectodermal-derived paraganglion tissue.1 These tumors are usually located along the vascular tree, especially near the carotid body, the jugulotympanic body, or the mediastinal vessels.2
While these low-grade tumors often occur in the superior or inferior para-aortic regions, there are reports of their presence in virtually every organ. However, the existence of paragangliomas as primary tumors of the lung remains controversial. Many cases reported as primary pulmonary paragangliomas (PPP) are not altogether convincing. This is because completely unrelated lesions, currently classified as meningoendothelial nodules, were previously known as chemodectomas and were classified as paragangliomas in the 1981 World Health Organization classification of tumors.1
Both solitary and diffuse PPP have been encountered in the lungs. Until recently they were classified as either pulmonary chemodectomas or carotid-body-like tumors of the lung.4 The solitary form of this tumor is rare.5 Heppleston first described PPP in 1958, and according to Aubertine, until 2004, only 23 cases were confirmed.1
We present a case of PPP with histologic and immunohistochemical findings and a critical review of the literature.
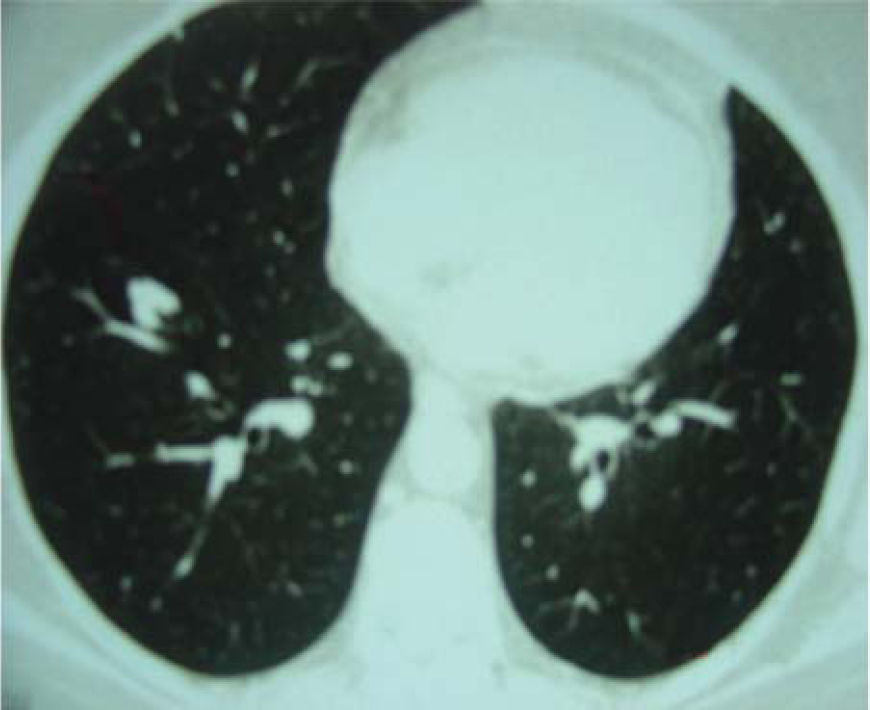
A 60-year-old woman with no previous symptoms presented to the Department of Thoracic Surgery of the Hospital do Câncer AC Camargo, where a routine chest x-ray revealed a pulmonary nodule. She was in good health and had quit cigarette smoking 10 years earlier. The clinical examination showed no remarkable findings, and the laboratory tests were normal. Computed tomography of the chest showed in the right lower lobe a nodule 12.0 mm in diameter with a lobulated margin (Figure 1). No significant alterations were observed in other organs. The patient underwent a right thoracotomy, and a segmentectomy was necessary for complete resection. Frozen section evaluation showed an irregular nodule with a smooth surface and ground glass opacity, consistent with a benign lesion. The postoperative course was uneventful, and she was discharged on the fourth day after surgery in good clinical condition.
")
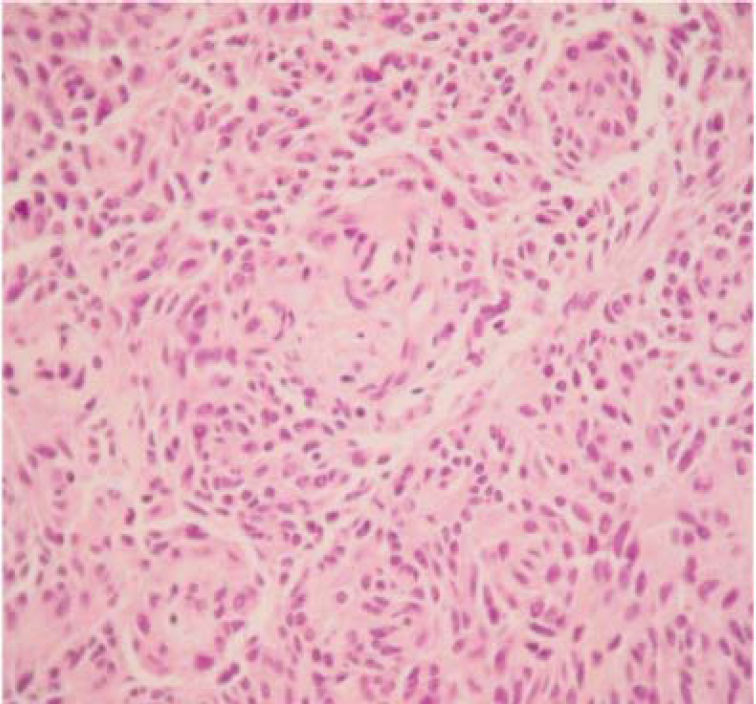
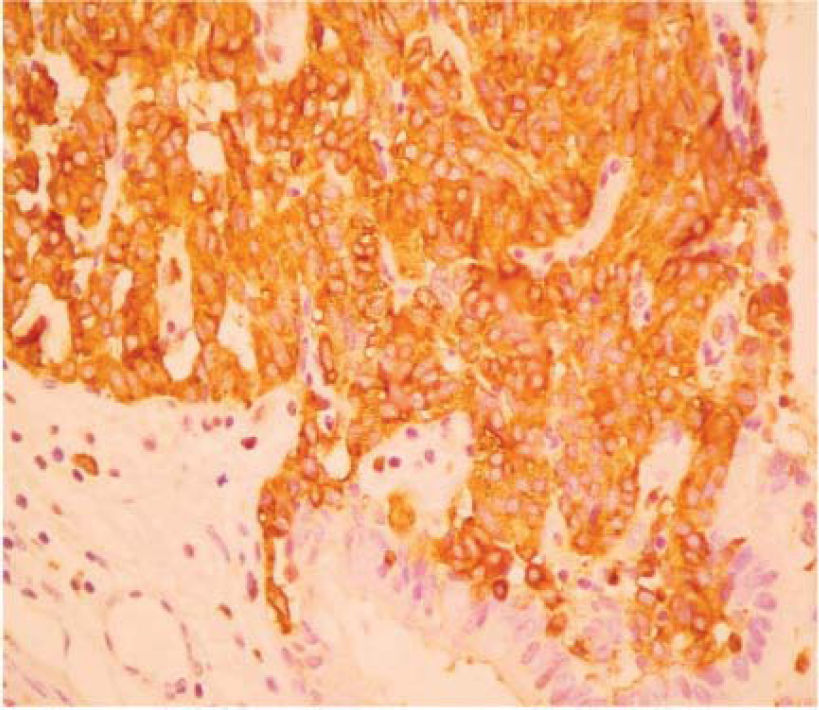
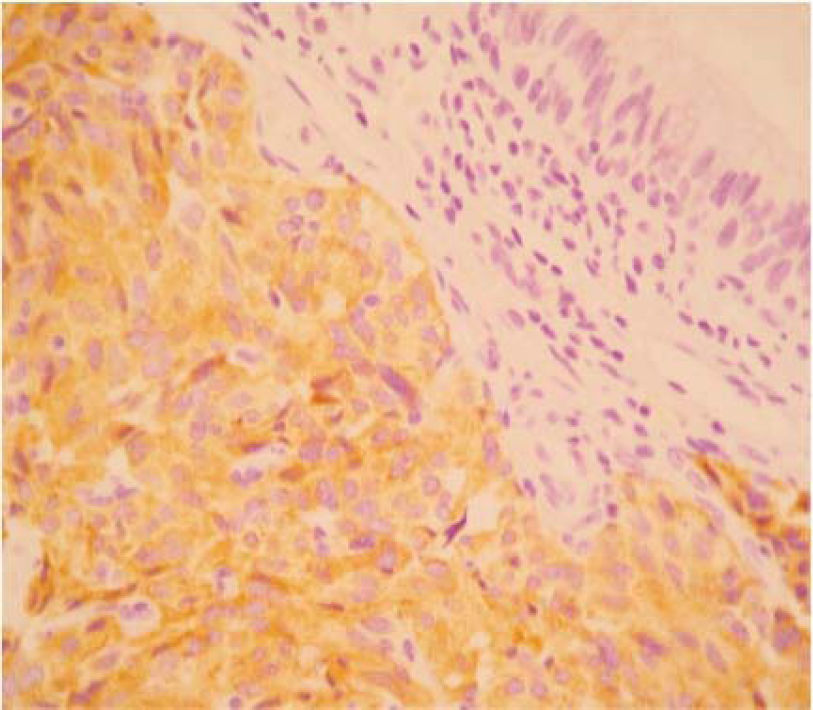
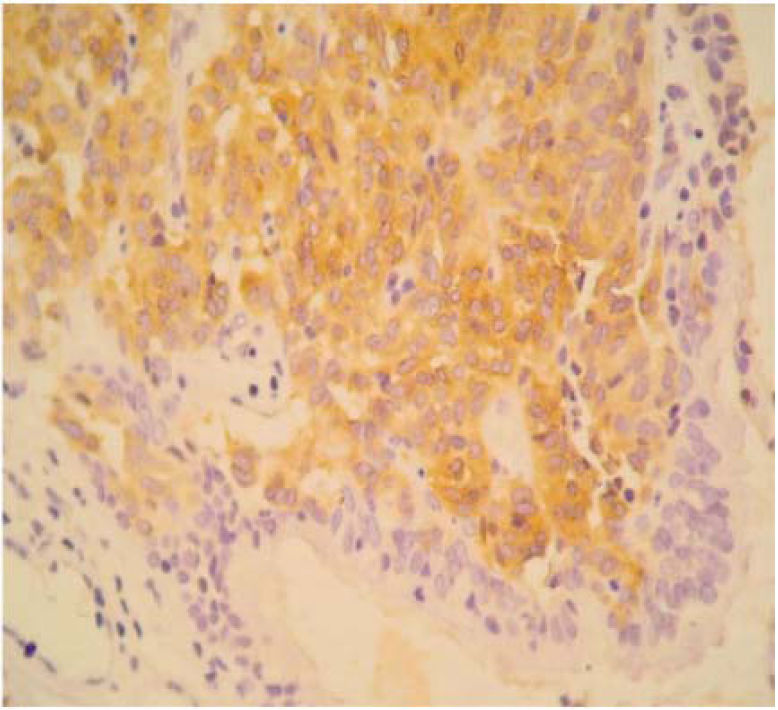
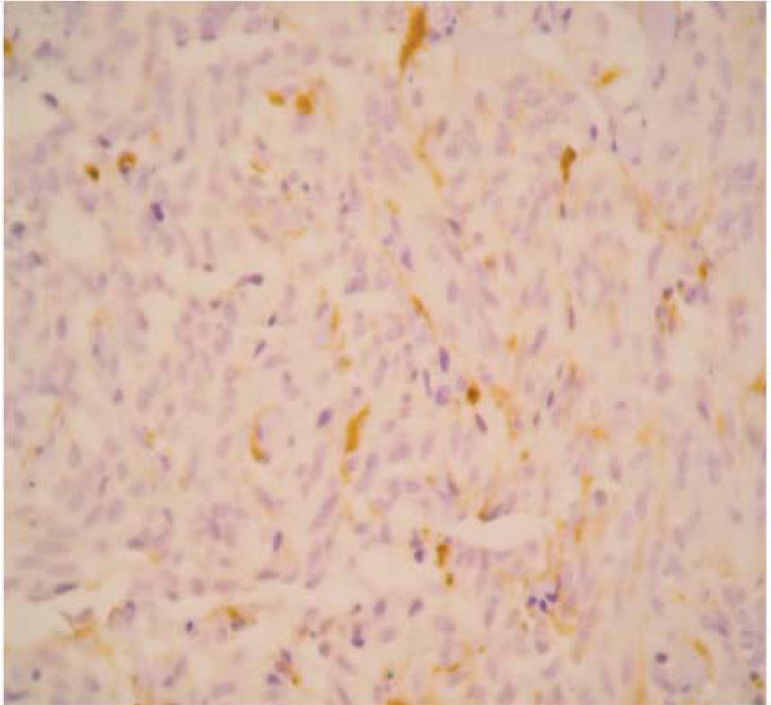
The final pathology report showed a nodular, well circumscribed, lobulated tumor located in the bronchial submucosa. The tumor was surrounded by bronchial cartilage with no infiltration. Histologically, the tumor was hypercellular, arranged in an anastomosing trabecular or nesting pattern (zellballen) within an arcuate vascular network (Figure 2). It was composed of nests of ovoid or polygonal cells, with abundant acidophilic cytoplasm and round-to-oval and occasional spindle nuclei with speckled chromatin. Conspicuous nucleoli were occasionally visible. Mitosis was absent. Nests of cells were surrounded by stellate spindle sustentacular cells and thin-walled vascular channels. Chief cells were intensely positive for neuroendocrine markers such as chromogranin A (Figure 3), synaptophysin, and neuron-specific enolase (NSE) (Figures 4 and 5). Sustentacular cells were positive for S-100 protein (Figure 6), but negative for cytokeratins (AE1/AE3, CK7, CK20, CK8). Expression of thyroid transcription factor-1 was not observed. All these findings were consistent with pulmonary paraganglioma.
")
")
")
")
")
Six months of follow-up produced no evidence of recurrence, and no further treatment was required.
After the first case of a solitary PPP reported by Heppleston, at least 23 cases have been described. This case report contributes a well-documented example of PPP. The presented neoplasm meets the Armed Forces Institute of Pathology's suggested morphologic criteria for the diagnosis of paraganglioma. These include a diffuse zellballen pattern throughout the tumor, the absence of classic carcinoid tumor architecture, the presence of cytoplasmic vacuoles distinctive of paragangliomas, a negative immunohistochemical staining for cytokeratin, and the exclusion of an extrapulmonary paraganglioma or pheochromocytoma.6
Primary pulmonary paraganglioma of the lung is distinct from the so-called multiple chemodectoma in its site of origin, growth pattern, and ultrastructure. The presence of neurosecretory granules does not permit a distinction between paraganglioma and bronchial carcinoid tumors.1,5 Some previous case reports based the diagnosis on the presence of sustentacular cells, because their presence was originally thought to be a specific diagnostic feature of paragangliomas. However, many studies have reported S-100 protein positive stellate cells resembling sustentacular cells in bronchial carcinoids in up to 76% of cases. While there is some ongoing debate as to whether these S-100 protein positive cells are mesenchymal or neuroendocrine in origin, it is generally agreed that they are not a distinguishing feature of paragangliomas. As the Armed Forces Institute of Pathology criteria suggest, cytokeratin can aid in the distinction between a PPP and a carcinoid, but is not absolutely diagnostic. As expected, nearly 100% of bronchial carcinoids, despite their degree of differentiation, will be positive for cytokeratins. Some previous cases had positive staining for cytokeratin; therefore, they are more consistent with a carcinoid tumor.8 The most recent World Health Organization classification has corrected the confusion by defining the meningoendothelial nodule and paraganglioma as entirely separate entities. In addition, the morphologically distinct gangliocytic paraganglioma should not be confused with a paraganglioma.
The grouping of tumors of the paraganglion system, with a subclassification based upon the specific paraganglionic tissue (branchiomeric, intravagal, aortico-sympathetic, or visceral autonomic), serves the useful purpose of combining tumors having a similar anatomic distribution, innervation, and microscopic structure. A further division of paraganglion tumors into functional or nonfunctional provides a clinically valuable distinction.4,6 Two forms of pulmonary paraganglia have been described. The first consists of multiple, minute pulmonary paragangliomas found in approximately 3% of the normal population at autopsy.7 Some authors suggest that these minute lesions represent ischemic stimulation of pre-existing paraganglionic cells resulting from pulmonary vascular obstruction. Association with pulmonary thromboemboli has been described in the adjacent blood vessel.1–4 Clinical symptoms and malignant activity have not been noted. The second form of pulmonary paraganglioma is a large solid tumor in the lung parenchyma, which was first reported in 1958 by Heppleston. As reported by Aron in 1977, all the tumors were removed surgically, with most of them located subpleurally.4 There was no evidence of local invasion of any other structure except in 1 case, in which the tumor had invaded the brachial plexus. Also important is the fact that in the lung, metastatic paraganglioma is more common than PPP.3
Until 1995, 21 cases had been reported and reviewed by Skot et al.3 The majority of these patients were asymptomatic, but 3 patients had hypertension, perhaps for other reasons. Seventeen of the tumors were located in the right lung and 5 in the left lung. Eighteen tumors were grossly and histologically benign. One tumor showed invasive growth and penetration of the capsule but no other signs of malignancy. In 1 case, which was the largest reported tumor, there was local invasion of the apex of the pleural cavity and of the brachial plexus, with some tumor tissue attached to the brachial plexus that was left behind at the operation.4 Areas of hemorrhage and necrosis in this tumor were described, but no lymph node metastases were found. Our case also showed no necrosis or mitosis, and no lymph nodes were found during surgery. Malignant PPP is distinctly uncommon, with only 1 case having been reported in a 49-year-old woman.2 She had lymph node metastases, but there was no evidence of distant metastases or any extrapulmonary involvement.
The distinction between benign and malignant paraganglioma is based on a low metastatic rate with a biologic behavior similar to that of a typical carcinoid. The differential diagnosis may be difficult on a purely morphological basis. In several studies, attempts have been made to distinguish between benign and malignant cases by immunohistochemical investigations. Some authors have found a reduced number of the S-100 protein-immunoreactive sustentacular cells 4,8 but these results were not confirmed in other studies. It seems that decreased immunoreactivity against NSE is found in malignant paragangliomas.
With the present experience, the acceptable treatment appears to vary from enucleation, wedge resection, and segmentectomy if the tumor is small, and lobectomy if the tumor is large or location permits. As in our case, surgical treatment seems to be sufficient, and extensive resections are not warranted.3,4
Our findings confirm the existence of PPP and show the importance of the differential diagnosis between paragangliomas and lung tumors such as typical carcinoids.