



The aim of the present study was to assess the effects of rosuvastatin on renal injury and inflammation in a model of nitric oxide deficiency.
METHODS:Male Wistar rats were randomly divided into four groups (n = 10/group) and treated for 28 days with saline (CTRL); 30 mg/kg/day L-NAME (L-name); L-NAME and 20 mg/kg/day rosuvastatin (L-name+ROS-20); or L-NAME and 2 mg/kg/day rosuvastatin (L-name+ROS-2). Systolic blood pressure was measured by plethysmography in the central artery of the tail. The serum total cholesterol, triglycerides, alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, creatinine, nitric oxide, interleukin-6, and tumor necrosis factor alpha levels were analyzed. Urine samples were taken to measure the albumin:urinary creatinine ratio. Kidneys were sectioned and stained with hematoxylin/eosin and Masson's trichrome. Immunohistochemical analysis of the renal tissue was performed to detect macrophage infiltration of the glomeruli.
RESULTS:The systolic blood pressure was elevated in the L-name but not the L-name+rosuvastatin-20 and L-name+rosuvastatin-2 groups. The L-name group had a significantly reduced nitric oxide level and an increased interleukin-6 and tumor necrosis factor alpha level, albumin:urinary creatinine ratio and number of macrophages in the renal glomeruli. Rosuvastatin increased the nitric oxide level in the L-name+rosuvastatin-2 group and reduced the interleukin-6 and tumor necrosis factor alpha levels, glomerular macrophage number and albumin:urinary creatinine ratio in the L-name+rosuvastatin-20 and L-name+rosuvastatin-2 groups.
CONCLUSION:Rosuvastatin treatment reduced glomerular damage due to improvement in the inflammatory pattern independent of the systolic blood pressure and serum lipid level. These effects may lead to improvements in the treatment of kidney disease.
Statins, which are 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, are currently the most popular drugs for the management of dyslipidemia for the primary and secondary prevention of cardiovascular disease.1 Their widespread use is driven by a robust body of evidence that supports their efficacy in preventing cardiovascular events.2 Their benefits are derived both from reducing atherogenic lipoprotein levels (LDL-C) and from increasing antiatherogenic lipoproteins levels (HDL-C).3
In addition to modulating lipid levels, recent clinical and experimental studies have shown that statins have pleiotropic effects, including antiinflammatory, antiproliferative, and antithrombotic effects; attenuation of NADPH oxidase–mediated superoxide generation; and improvement of endothelial vasomotor function. All of these effects may influence cardiovascular outcomes, including hypertension, in high-risk patients.4 Statins reduce vascular inflammation, suppress extracellular matrix production, exhibit inhibitory effects on the Rho/Rho kinase pathway,5,6 and block small G protein isoprenylation, which is a pivotal step in the activation of Rho.
The kidney is a target organ of hypertension. Renal disease secondary to hypertension progressively develops, culminating in chronic renal failure with a loss of glomeruli and several morphological and quantitative alterations.7,8 The renal lesions caused by nitric oxide (NO) blockade are glomerulosclerosis, interstitial fibrosis, and microvascular lesions.9,10
The current chronic nephropathy treatments are limited to angiotensin-converting enzyme inhibitors and angiotensin receptor blockers.11 However, similar to observations in the cardiovascular system, increasing clinical12 and experimental13–17 evidence indicates that statins have beneficial effects in the course of progressive renal disease. In addition, statins influence intrarenal hemodynamics by reducing angiotensin and aldosterone levels.18
Recent studies have shown that chronic administration of L-arginine analogues such as L-nitro-arginine methyl ester (L-NAME) to rats induces dose-dependent systemic arterial hypertension by blocking endothelial nitric oxide synthase (eNOS) and, therefore, NO biosynthesis.19,20
The rat hypertension model induced by chronic inhibition of NO generation has been shown to be a useful tool for studying both the development and treatment of renal lesions resembling those found in human hypertension, a disease associated with early generalized impairment of endothelial function.21 This hypertension and renal disease model seems to be a suitable model to examine the lipid lowering–independent effects of statins because the administration of statins, even at high doses, does not alter serum cholesterol levels in normal rats.22–23
The aim of this study was to assess the effects of rosuvastatin on renal injury and inflammation in a model of nitric oxide deficiency.
METHODS AND MATERIALAnimalsAll animals (n = 40; 6-week-old male Wistar rats) were maintained in standard cages at room temperature (22±3°C) and 60–70% humidity with a 12 h dark/12 h light cycle and ad libitum food (Nuvilab, Paraná, Brazil) and water. This diet contained 22.5% crude protein and 0.27% sodium. Twelve hours before each experiment, the animals received only water to avoid interference with absorption of the treatments. The rats were euthanized with 50 mg/kg i.p. sodium pentobarbital. Systolic blood pressure was measured weekly in conscious rats using noninvasive tail-cuff plethysmography (Letica 5001, Panlab, Spain) at baseline and at the end of each week of treatment. The experiment was conducted for four weeks. This study was conducted in accordance with guidelines set forth by the Brazilian Association for Laboratory Animal Science (COBEA) and has been approved by the Research Commission for Ethics and Animal Experimentation of UFJF.
TreatmentsThe rats were randomly divided into four groups (n = 10/group) and treated as follows: a control group (CTRL); 30 mg/Kg/day L-NAME hydrochloride (Sigma, St. Louis, USA) diluted in the drinking water (L-name); L-NAME+20 mg/kg/day rosuvastatin (Crestor, AstraZeneca, Brazil) delivered by gavage (L-name+ROS-20); and L-NAME+2 mg/kg/day rosuvastatin (L-name+ROS-2) delivered by gavage. The L-NAME in the drinking water was at a concentration of 500 mg/l, and each non-control rat received 30 mg/kg/day of L-NAME for 28 days.
Biochemical parametersBlood samples were drawn by cardiac puncture from the right ventricle to measure the serum levels of total cholesterol (TC), triglycerides (Tg), aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and creatinine (Cr) (Cobas Mira, Roche). The serum level of NO was assessed using the Griess method.24 The levels of interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) were measured using enzyme-linked immunosorbent assay (ELISA) (Opt, BD, USA) according to the manufacturer's instructions. Urine was obtained by bladder puncture to establish the urine albumin: creatinine ratio (Biotecnica kit, Cobas Mira, Roche). Analyses were performed in duplicate, and samples were stored at -80°C until analysis.
Histological analysisAfter euthanasia, a fixative solution (1.27 mol/l of freshly prepared formaldehyde in 0.1 M phosphate buffer; pH 7.4) was perfused through the vascular system using a catheter placed into the left ventricle until the body was rigid. The kidneys were removed, cut into two halves, placed in 10% formaldehyde for 48 h at room temperature, dehydrated, and embedded in paraffin for histological analysis. Sections (3 μm thick) were cut and stained with hematoxylin/eosin for morphological analysis or Masson's trichrome to highlight collagen fibers. Histological analysis was performed using a light microscope (Olympus BX51). Images were captured (Color Coolsnap-Pro, Media Cybernetics) and analyzed using Image Pro-Plus software (Version 6.0, Media Cybernetics, Silver Spring, USA). The sections were evaluated by a pathologist without prior knowledge of the groups to which the animals belonged. Tubules (to study tubular dilation), blood vessels (to identify intimal fibrosis, microthrombosis and fibrinoid necrosis), glomeruli (to identify glomerulosclerosis and glomerular atrophy), and the interstitial compartment (to detect changes in collagen and inflammatory cells (400×)) were analyzed morphologically to identify the range of changes. The glomerular surface area (μm2) was morphometrically analyzed in at least 50 glomeruli per animal.25
Immunohistochemical analysisImmunohistochemical analysis of renal tissues to detect the number of macrophages in 100 random glomeruli per animal was performed on paraffin-embedded sections as described by Hartner et al.26 using antibodies against macrophages/monocytes (Mouse anti-rat CD68; Serotec). The percentage of glomeruli with macrophages was determined by counting 100 random clusters from nearly 20 microscopic fields, in accordance with Pomaro et al.,27 and was determined by a pathologist without prior knowledge of the groups to which the animals belonged.
Statistical analysisThe results are presented as the mean ± standard error (SEM). The data were all normally distributed in each group (Kolmogorov-Smirnov test, p>0.05). Statistical analysis between groups was performed using an analysis of variance (one-way ANOVA) followed by Tukey's test when the data were homoscedastic. If the data were heteroscedastic, the nonparametric Kruskal-Wallis test followed by the Tamhane test was performed. A p-value less than 0.05 was considered significant. Analyses were conducted and graphs were created using GraphPad Prism Version 5.01 for Windows (GraphPad Software, San Diego, CA, USA) and Statistical Package for Social Sciences Version 13.0 (SPSS, Chicago, IL, USA).
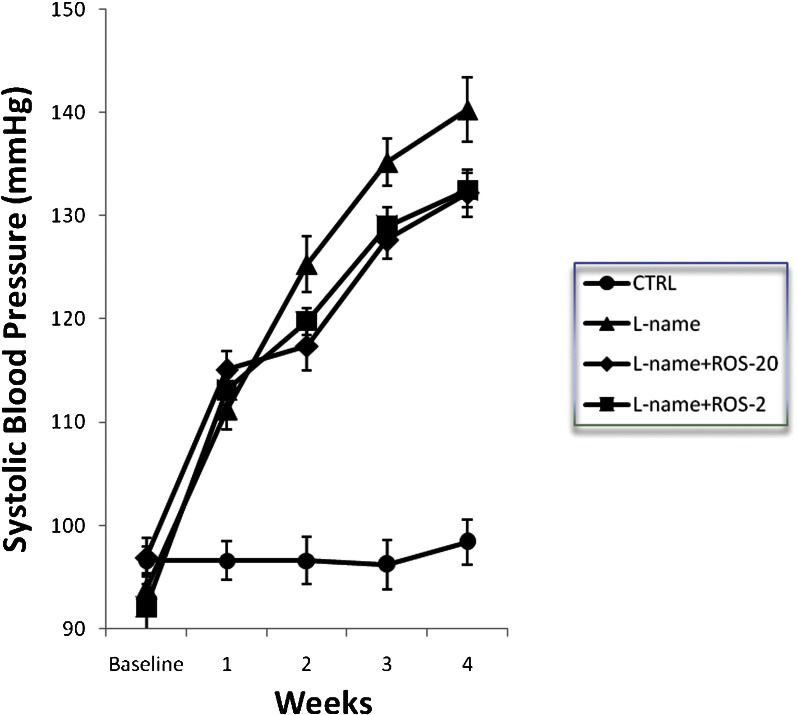
RESULTSSystolic blood pressure analysisThe initial systolic blood pressure (SBP) was similar among the groups. At the end of the first week of treatment with L-NAME, all L-NAME-treated groups had an elevated SBP compared with the control group (p<0.0001 for all of the groups). This hypertension was observed for all L-NAME-treated groups throughout the experiment (Figure 1; p<0.0001 for all groups).
 had significantly increased systolic blood pressures when compared with the CTRL group (p<0.0001).")
Systolic blood pressure over the entire experimental period in control and treated rats. The data are expressed as the mean ± SEM for = 10 rats/group. After the first week, all of the treated groups (L-name, L-name+ROS-20, L-name+ROS-2) had significantly increased systolic blood pressures when compared with the CTRL group (p<0.0001).
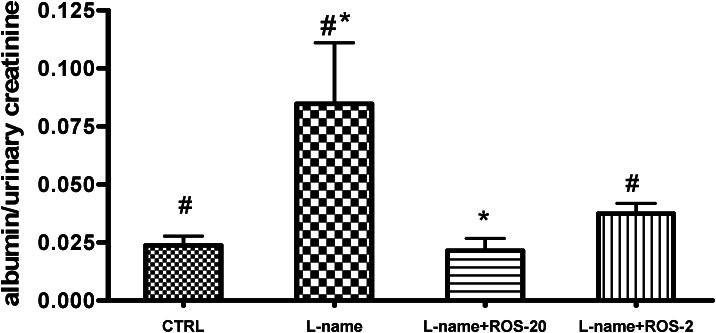
The experimental groups had similar TC, Tg, AST, ALT, ALP, and Cr levels (Table 1). Compared with the control, the L-name group had increased serum levels of IL-6 (p<0.001) and TNF-α (p<0.01). However, when compared with the L-name group, treatment with 2 or 20 mg/kg/day rosuvastatin inhibited the L-NAME-induced increase in the serum IL-6 (p<0.001 for both groups) and TNF-α levels (L-name+ROS-20, p = 0.030; L-name+ROS-2, p = 0.028). The L-name group showed a significant reduction in the NO level compared with the control group (p = 0.01). Only the L-name+ROS-2 group showed significantly higher levels of NO compared with the L-name group (p = 0.047; Table 2). The L-name group showed an increase in the urinary albumin:creatinine ratio compared with the CTRL group (p = 0.023). Rosuvastatin at both tested doses (2 and 20 mg/kg/day) lowered the urinary albumin:creatinine ratio compared with the L-name group (p = 0.008 and p = 0.047, respectively) (Figure 2).
Biochemical parameters of the experimental groups at the end of the experiment.
| Parameter | CTRL | L-name | L-name+ROS-20 | L-name+ROS-2 |
|---|---|---|---|---|
| n | 10 | 10 | 10 | 10 |
| Serum creatinine, mg/dl | 1.1±0.04 | 0.9±0.03 | 1.0±0.09 | 1.0±0.07 |
| Total cholesterol, mg/dl | 71.0±6.3 | 74.1±4.4 | 82.5±7.0 | 72.8±3.9 |
| Triglycerides, mg/dl | 73.8±5.8 | 89.2±6.8 | 78.7±15.7 | 72.6±4.5 |
| Alanine aminotransferase, U/l | 250.3±45.1 | 222.4±7.8 | 227.3±13.1 | 186.1±13.2 |
| Aspartate aminotransferase, U/l | 102.3±12.1 | 112.5±3.5 | 116.0±4.0 | 99.2±2.4 |
| Alkaline phosphatase, U/l | 236.0±14.6 | 254.2±10.2 | 259.9±23.5 | 236.7±15.3 |
Data are expressed as the mean ± SEM. All groups had similar biochemical parameters when compared with the CTRL group.
Inflammatory parameters of the experimental groups at the end of the experiment.
| Group | IL-6 (pg/mL) | TNF-α (pg/mL) | NO (μM) |
|---|---|---|---|
| CTRL | 7.4±0.4∗ | 25.5±1.8# | 2.1±0.5# |
| L-name | 25.3±2.0 | 35.3±4.7 | 0.5±0.1 |
| L-name+ROS-20 | 8.7±0.1∗ | 26.1±1.4+ | 0.8±0.1 |
| L-name+ROS-2 | 10.2±0.5∗ | 25.4±1.2+ | 2.3±0.6# |
The results are expressed as the mean ± SEM. n = 10 rats/group.
NO: serum nitric oxide level; IL-6: interleukin-6; TNF-α: tumor necrosis factor-alpha.

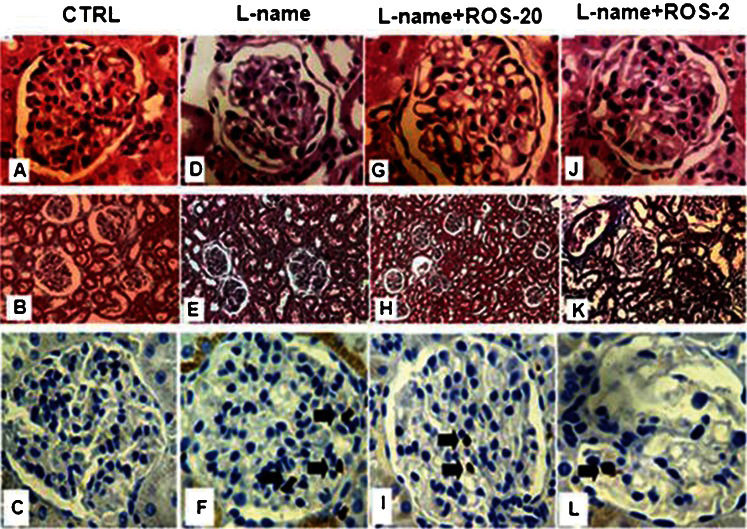
Histological evaluation following staining with hematoxylin/eosin (Figure 3 A,D,G,F) or Masson's trichrome (Figure 3 B,E,H,K) revealed no detectable renal pathology in any of the groups in all of the renal compartments. The cross-sectional glomerular areas were similar among all of the experimental groups (Table 3).
, cortical sections stained with Masson")
Histological and immunohistochemical analysis. Representative glomerular sections stained with hematoxylin/eosin (A,D,G,J; 400×), cortical sections stained with Masson's trichrome (B,E,H,K; 20×) and immunohistochemical analyses of glomerular macrophages (C,F,I,L) in each group of rats (n = 10/group). These analyses did not detect changes in the glomerular, vascular, tubular or interstitial compartments. Glomerular macrophages were most abundant in the L-name group (arrow).
Glomerular macrophage infiltration and the cross-sectional glomerular area of the kidneys of animals at the end of the experiment.
| Group | Glomerular macrophage (%) | Cross-sectional glomerular area (μm2) |
|---|---|---|
| CTRL | 39±3∗ | 3833±61 |
| L-name | 107±4 | 3673±64 |
| L-name+ROS-20 | 56±4∗ | 3688±83 |
| L-name+ROS-2 | 45±6∗ | 3911±75 |
The results are expressed as the mean ± SEM. n = 10 rats/group. The number of macrophages was analyzed in 100 random glomeruli per animal. The glomerular surface area was morphometrically analyzed in at least 50 glomeruli per animal.
The L-name group demonstrated an increased number of macrophages in the renal glomeruli compared with the CTRL group (p<0.0001). The L-name+ROS-20 and L-name+ROS-2 groups demonstrated decreased infiltration of macrophages into the glomeruli compared with the L-name group (p<0.0001) (Figure 3 C,F,I,L and Table 3).
DISCUSSIONChronic nitric oxide synthesis inhibition induced by the treatment of rats with L-NAME results in endothelial dysfunction, vascular hypertrophy, cardiac fibrosis, atherosclerosis, perivascular inflammation, renal failure, and increased vascular responses to adrenergic stimuli. Several other factors, including Renin-angiotensin system, endothelial constrictor factors, arterial remodeling and the sympathetic nervous system are involved in these effects.19,28 Additionally, this experimental model is characterized by easy handling and low animal mortality.
The dose of 30 mg/kg/day of L-NAME was sufficient to induce arterial hypertension,29 and the dose of 20 mg/kg/day of rosuvastatin used in our study is known to have beneficial effects on endothelial dysfunction and systemic and regional hemodynamics.23 At 2 mg/kg/day, rosuvastatin has been shown to significantly increase eNOS mRNA and protein expression and decrease iNOS mRNA and protein expression and nitrite production after ischemia reperfusion.30
The L-name group developed hypertension after the end of the first induction week. At the end of the experiment, there was a decrease in the serum NO level and an increase in inflammatory parameters (the serum IL-6 and TNF-α levels), macrophage infiltration into the glomeruli and proteinuria.
The effects of statins on blood pressure can be dose dependent in some animal models, such as spontaneously hypertensive31 and angiotensin II–dependent rats.32 Our results did not demonstrate a dose-dependent reduction in blood pressure following treatment with rosuvastatin (2 or 20 mg/kg/day). A similar finding was reported by Susic et al.23
In rats, serum lipid levels are usually low, and statins do not change this profile. Therefore, rats are an excellent model to study the pleiotropic effects of statins.23 In our study, treatment with rosuvastatin did not alter the serum Tg, TC, ALT, AST, ALP, or Cr levels, which indicates that rosuvastatin exerted lipid lowering–independent effects. Furthermore, in our study, the serum ALT, AST, and ALP levels indicated no liver toxicity.
Biochemical markers, particularly markers of vascular inflammation, such as high-sensitivity C-reactive protein (hs-CRP), have been suggested to be predictive of cardiovascular events.33 The primary proinflammatory cytokines TNF-α and IL-6 are the main inducers of hs-CRP. Furthermore, according to Navarro and Mora-Fernandez,34 TNF-α and IL-6 have been associated with significant direct renal effects.
In this study, treatment with rosuvastatin (2 or 20 mg/kg/day) reduced the serum IL-6 and TNF-α levels. Experimental and clinical studies have demonstrated the pathogenic role of TNF-α in the development of renal injury and the potential benefit of modulating TNF-α activity as a therapeutic target in diverse renal diseases.35
IL-6 has been correlated with an increased width of the glomerular basement membrane.35 It also enhances fibronectin expression, affects extracellular matrix dynamics at both the mesangial and podocyte levels, stimulates mesangial cell proliferation, and increases endothelial permeability.36,37 IL-6 plays an important role in vascular remodeling and has been reported to be a useful biomarker in predicting future cardiovascular events.38
TNF-α is cytotoxic to glomerular, mesangial and epithelial cells, and it is able to induce direct renal damage.39 Furthermore, TNF-α stimulates endothelial generation of reactive oxygen species by activating the NADPH oxidase subunits gp91 phox, NOX-1, p47phox, and p22phox. TNF-α also activates the transcription of NF-κB, which regulates the expression of genes involved in inflammation, oxidative stress and endothelial dysfunction.11,38
Animals treated simultaneously with L-NAME and rosuvastatin (2 mg/kg/day; L-name+ROS-2) had an increased serum NO level and reduced L-NAME-induced inflammatory parameters. However, treatment with 20 mg/kg/day rosuvastatin (L-name+ROS-20) did not alter the serum NO levels compared with the L-name group. This finding could be because a lower dose (2 mg/kg/day) of rosuvastatin may increase the constitutive endothelial bioavailability of NO, which may not occur at higher doses (20 mg/kg/day). Di Napoli et al.30 evaluated the effects of rosuvastatin on reperfusion injury in rat hearts using 0.2, 2, and 20 mg/kg/day rosuvastatin. They found that 2 mg/kg/day rosuvastatin significantly increased the mRNA expression of constitutive endothelial NOS (eNOS) compared with the untreated group and reduced the mRNA expression of inducible NOS (iNOS). These effects were more evident at 2 mg/kg/day rosuvastatin than at 0.2 mg/kg/day rosuvastatin. However, at the highest dosage (20 mg/kg/day) there was a beneficial effects loss in the expression of eNOS and iNOS associated with increased mitochondrial damage and microcirculatory dysfunction.
The current treatment of chronic nephropathy is limited to angiotensin-converting enzyme inhibitors and angiotensin AT1 receptor blockers; however, evidence indicates that statins can be effective.11
Glomerulosclerosis, glomerular ischemia, and interstitial fibrosis were observed in the kidneys of nitric oxide–deficient rats fed a high-salt diet.40 Changes in glomerular and vascular densities were also observed in this model using stereological analysis.41 The BPs achieved in the animals used in these previous studies were 212 and 200–225 mmHg, respectively. However, Lecian et al.42 found no histological abnormalities in the kidneys of rats treated with a nitric oxide synthesis inhibitor, and these animals had a BP of 175 mmHg. In our study, the BP of the hypertensive group only reached 140 mmHg, which may explain the absence of histological abnormalities.
Statins inhibit several intracellular signaling pathways (Rho/RhoA and MAPK pathways) activated by angiotensin II that are involved in the regulation of profibrotic factors, connective tissue growth factor and the deposition of extracellular matrix components, which are all modulated by redox processes.43 Our data demonstrate that there was a significant increase in the accumulation of macrophages in the glomeruli and proteinuria in L-NAME-treated rats. Rosuvastatin was effective in reducing glomerular macrophage infiltration, which is indicative of an improved inflammatory pattern; however, this effect was most evident at a dose of 2 mg/kg/day. Additionally, rosuvastatin inhibited proteinuria at both of the tested doses (2 or 20 mg/kg/day).
CONCLUSIONSIn conclusion, rosuvastatin reduces the serum levels of IL-6 and TNF-α, reduces glomerular macrophage infiltration and prevents proteinuria in this experimental model. All of the effects of rosuvastatin were independent of its lipid-lowering effects. Future mechanistic studies will examine the molecular mechanisms involved in these effects. Our data suggest that rosuvastatin may act beneficially in preventing proteinuria and glomerular inflammation in a model of nitric oxide deficiency.