



The aim of this study was to evaluate overall genetic damage induced by total sleep deprivation in obese, female Zucker rats of differing ages.
METHODLean and obese Zucker rats at 3, 6, and 15 months old were randomly distributed into two groups for each age group: home-cage control and sleep-deprived (N = 5/group). The sleep-deprived groups were deprived sleep by gentle handling for 6 hours, whereas the home-cage control group was allowed to remain undisturbed in their home-cage. At the end of the sleep deprivation period, or after an equivalent amount of time for the home-cage control groups, the rats were brought to an adjacent room and decapitated. The blood, brain, and liver tissue were collected and stored individually to evaluate DNA damage.
RESULTSSignificant genetic damage was observed only in 15-month-old rats. Genetic damage was present in the liver cells from sleep-deprived obese rats compared with lean rats in the same condition. Sleep deprivation was associated with genetic damage in brain cells regardless of obesity status. DNA damage was observed in the peripheral blood cells regardless of sleep condition or obesity status.
CONCLUSIONTaken together, these results suggest that obesity was associated with genetic damage in liver cells, whereas sleep deprivation was associated with DNA damage in brain cells. These results also indicate that there is no synergistic effect of these noxious conditions on the overall level of genetic damage. In addition, the level of DNA damage was significantly higher in 15-month-old rats compared to younger rats.
Factors such as sleep curtailment, obesity, and age can exert a wide variety of negative effects on the immune system and induce pathological processes such as genomic damage (1,2). Moreover, these factors are able to reduce the body's capacity to repair DNA damage, increase the cellular sensitivity to oxidative stress, and promote cell death (3). All of this evidence demonstrates that the repair of oxidative damage in DNA is a dynamic and highly regulated process. When DNA repair is inhibited, cells age prematurely and die more easily (4).
One useful method for quantifying DNA damage is the alkaline comet assay (single-cell gel electrophoresis) (5,6). Because of the simplicity and sensitivity of this method, the comet assay has rapidly gained acceptance as a genotoxicity assay (7). The basic principle of the comet assay is the migration of DNA in an agarose matrix under electrophoretic conditions. When viewed under a microscope, a damaged cell has the appearance of a comet, with a head (nuclear region) and a tail that contains DNA fragments or strands migrating towards the anode (8). Previous studies have demonstrated that the comet assay is a suitable tool to investigate genotoxicity in vivo (9,10). The extent of the comet is directly related to the level of DNA damage (11,12). Hence, the comet assay is a useful technique to detect DNA strand breaks in multiple organs and under different conditions, such as sleep deprivation (SD), disease progression, drug abuse, etc. (13-17). For example, we have used this technique to detect DNA breakage in vivo in medium-term carcinogenesis assays and parasitic infections and following exposure to xenobiotics that are present in the environment (9,15,18).
In recent years, increasing attention has been given to the genetic changes that are caused by sleep loss in different contexts of SD (16,19). Recently, we demonstrated that SD induces genetic damage in the blood and brains cells of male rats (16), indicating that lack of sleep leads to severe molecular damage (19). In humans, the shortening of sleep can occur due to various factors, such as social life, artificial light, shift work, or sleep disturbances. Indeed, sleep loss is heightened in societies that have a frenzied lifestyle. Additionally, most sleep disorders, such as sleep apnea and insomnia, lead to sleep deprivation, which disrupts vital biological processes that are necessary for physical health (20). SD and obesity have been linked with comorbidities associated with morbid obesity rather than being causal factors (20,21). However, to the best of our knowledge, no study has investigated the influence of these risk factors on the genome at the single-cell level in this rat strain. Thus, the aim of this investigation was to evaluate overall genetic damage induced by sleep loss in obese female Zucker rats at different time points during their life span.
MATERIAL AND METHODSAnimalsThe experiments in this study were performed on female lean and obese adult Zucker rats (3, 6, and 15 months old). The animals were provided by the Centro de Desenvolvimento de Modelos Experimentais para Medicina e Biologia (CEDEME) of the Universidade Federal de São Paulo. The animals were housed in groups of three in standard polypropylene cages in a room that was maintained at 22°C with a 12:12 h light-dark cycle (lights on at 7 am) and were given free access to food and water. The rats used in this study were maintained and treated in accordance with the guidelines established by the Ethical and Practical Principles for the Use of Laboratory Animals (22). All animal procedures were approved by the University Ethics Committee (Protocol #1268/08). Female rats were chosen because women have been shown to be frequently exposed to stressful events (12) and because their mortality is often increased prior to menopause (11).
Obese Zucker rats are frequently used as a model for early-onset, hyperplastic-hypertrophic obesity (23). Although Zucker rats cannot serve as an exact model of the human condition, these animals have provided important insights into the causes of human obesity and the possible consequences associated with this condition (24). Obesity in this strain is transmitted as a Mendelian autosomal recessive trait. Homozygous animals have been shown to accumulate fat progressively beginning at the 5th week of life (25) and demonstrate physical hypoactivity and hyperphagia. In addition, homozygous animals with access to food ad libitum will exhibit hyperinsulinemia (25,26), insulin resistance, hyperlipidemia (24,27), and hypothermia (27,28).
Total Sleep DeprivationAnimals in the SD group were subjected to a single episode of continuous SD during the first 6 hours of the light phase (7 am to 1 pm) using a gentle handling method. The SD was performed by introducing an object (a pick) into the cage and tapping the cages whenever the animals appeared drowsy. The animals were not disturbed during feeding or drinking (29). We chose this method rather than the platform technique because obese animals may have had difficulty standing on the narrow platforms inside the water tank, which are used in the conventional platform method.
Experimental DesignLean and obese Zucker rats (3, 6, and 15 months old) were randomly assigned to either the control (CTRL) or the sleep-deprived (SD) group for a total of 12 groups (five animals/group). The experiments were conducted while the female rats were in the diestrus phase of their estrous cycle. We elected to use rats only during diestrus because this phase was previously shown to be particularly affected by SD (30). The SD groups were handled gently for 6 hours, whereas the CTRL group was allowed to sleep in an adjacent room. The animals were sacrificed immediately after the SD period.
Sample CollectionAt the end of the SD period, or after an equivalent amount of time for the CTRL groups, the animals were brought to an adjacent room and were euthanized by decapitation between 1 and 3 pm. Blood was collected in sterile tubes containing liquid EDTA, aliquots were removed, and cellular suspensions (∼10 µl) were used for the comet assay. In addition, central fragments from the kidney, heart, liver, and brain (prefrontal cortex) were also collected and minced in 0.9% NaCl in preparation for the comet assay. The sub-region of the prefrontal cortex was chosen based on results from our previous studies (16,17,19). The samples were maintained at -80°C until the assays were performed. All of the samples were handled in an identical manner.
Comet AssayThe protocol used to process the peripheral blood, kidney, liver, heart, and brain cells was a modified version of the protocol outlined by Tice et al. (31). Briefly, a volume of 5 μl of peripheral blood or of the cellular suspensions from the liver, heart, kidney, or prefrontal cortex was added to 120 μl of 0.5% low-melting-point agarose at 37°C. The low-melting-point agarose containing cells was then layered onto a slide pre-coated with 1.5% regular agarose and covered with a coverslip. Prior to electrophoresis, the slides were left in an alkaline buffer (pH>13) for 20 min and were then electrophoresed for 20 min at 0.7 V/cm and 300 mA on ice. After electrophoresis, the slides were neutralized in 0.4 M Tris-HCl (pH 7.5), fixed in absolute ethanol, and stored until they were analyzed on a fluorescent microscope at 400× magnification. Independent positive controls, which were cells from the peripheral blood, liver, and brain, were treated in vitro with 10 μM H2O2 (hydrogen peroxide) for 10 min at 4°C to ensure the reproducibility and sensitivity of the assay. All of the positive controls were made using cells from control lean rats. All of the samples from CTRL lean rats were treated randomly, and all analyses were performed blinded.
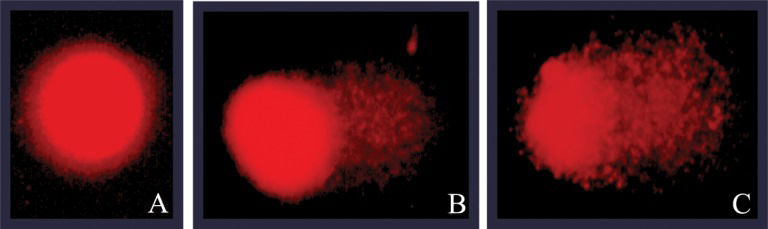
Analysis of Genotoxicity DataA total of 50 randomly captured comets per animal (25 cells from each slide) (18) were examined blindly by one expert observer. These comets were observed at 400× magnification using a fluorescent microscope (Olympus). The microscope was connected through a black and white camera to an image analysis system (Comet Assay II, Perceptive Instruments, Suffolk, Haverhill, UK). The instrument had been previously calibrated according to the manufacturer's instructions. The computerized image analysis system acquires images, computes the integrated intensity profiles for each cell, estimates the comet cell components, and evaluates the range of the derived parameters. Undamaged cells have an intact nucleus without a tail, whereas damaged cells have the appearance of a comet (Figure 1. To measure the level of DNA damage, the tail intensity (% migrated DNA) was considered (32).
Statistical Methods, a cell exposed to sleep deprivation (B), and an H2O2-treated cell (positive control) (C). DNA was stained with ethidium bromide; 40× magnification.")
The results obtained in the comet assay were evaluated statistically as recommended by Wiklund and Agurrel (33). Thus, the tail intensity data were log10-transformed and compared by one-way ANOVA followed by a post-hoc Dunn's test using Sigma Stat for Windows. According to the single-cell gel (comet) assay expert group, comet assay data should be log-transformed prior to statistical tests because they are not normally distributed (33). Body weight was analyzed using Student's t-test for independent samples comparing lean and obese rats within each age group. The level of significance was set at α = 0.05 for the experiments in this study.
RESULTSAs expected, the adult obese rats had significant increases in body weight compared to the lean group at 3 months old (404.0±35.8 vs. 205.3±16.2, p≤0.001), 6 months old (544.3±56.6 vs. 241.4±23.4, p≤0.001), and 15 months old (747.0±42.3 vs. 259.2±45.6, p≤0.001). However, the mean body weight of the obese group was not significantly different for the SD condition at any age compared to the CTRL condition.
No statistically significant differences were identified in the blood cells of the obese rats during the younger phases of life. In addition, SD was not able to induce DNA breakage at these ages. Nevertheless, DNA migration was observed in rats that were 15 months old, irrespective of their obesity status (Table 1. No significant differences were observed among the groups in the kidney or the heart. For the liver, DNA migration was observed in the liver cells of the SD obese rats at every age evaluated. SD alone was not able to induce genetic damage in the liver, as shown in Table 2.
DNA damage (expressed as tail moment data) in the peripheral blood from lean and obese Zucker rats that underwent sleep deprivation.
| Age | DNA damage (tail moment) | ||||
|---|---|---|---|---|---|
| Control | Sleep Deprivation | Positive control1 | |||
| Lean | Obese | Lean | Obese | ||
| 3 months | |||||
| 0.7 | 0.2 | 0.1 | 1.0 | 3.5 | |
| 0.9 | 1.0 | 0.6 | 0.7 | 2.3 | |
| 0.5 | 0.8 | 0.5 | 0.8 | 2.4 | |
| 0.5 | 0.3 | 0.9 | 0.5 | 5.2 | |
| 0.8 | 0.5 | 0.2 | 0.2 | 3.7 | |
| Mean±S.D. | 0.7±0.2 | 0.5±0.3 | 0.5±0.2 | 0.6±0.3 | 3.4±1# |
| 6 months | |||||
| 0.9 | 0.8 | 0.5 | 1.4 | 3.5 | |
| 1.3 | 0.2 | 0.8 | 0.6 | 2.3 | |
| 0.7 | 0.7 | 0.3 | 0.9 | 2.4 | |
| 0.5 | 0.4 | 0.2 | 1.4 | 5.2 | |
| 1.4 | 1.2 | 0.6 | 1.9 | 3.7 | |
| Mean±S.D. | 0.9±0.4 | 0.6±0.4 | 0.4±0.2 | 1.2±0.5 | 3.4±1# |
| 15 months | |||||
| 2.7 | 1.8 | 1.5 | 2.8 | 3.5 | |
| 2.3 | 1.2 | 1.0 | 1.5 | 2.3 | |
| 1.5 | 2.2 | 0.5 | 0.9 | 2.4 | |
| 1.7 | 0.8 | 0.7 | 1.3 | 5.2 | |
| 1.0 | 1.4 | 1.4 | 2.0 | 3.7 | |
| Mean±S.D. | 1.8±0.5*) | 1.5±0.5*) | 1.0±0.4*) | 1.7±0.6*) | 3.4±1# |
DNA damage (expressed as tail moment data) in the livers of lean and obese Zucker rats that underwent sleep deprivation.
| Age | DNA damage (tail moment) | ||||
|---|---|---|---|---|---|
| Control | Sleep Deprivation | Positive control1 | |||
| Lean | Obese | Lean | Obese | ||
| 3 months | |||||
| 1.3 | 1.7 | 0.3 | 1.7 | 3.3 | |
| 0.5 | 0.6 | 0.9 | 1.4 | 6.7 | |
| 0.7 | 1.5 | 0.4 | 1.9 | 4.5 | |
| 0.4 | 1.8 | 0.7 | 0.8 | 5.8 | |
| 0.4 | 1.6 | 0.2 | 2.3 | 7.0 | |
| Mean±S.D. | 0.7±0.4 | 1.5±0.5 | 0.5±0.3 | 1.6±0.5¥ | 5.4±1.5# |
| 6 months | |||||
| 0.6 | 2.4 | 0.8 | 3.0 | 3.3 | |
| 1.2 | 1.0 | 0.8 | 3.2 | 6.7 | |
| 0.4 | 1.4 | 0.6 | 2.1 | 4.5 | |
| 0.5 | 2.0 | 0.5 | 1.8 | 5.8 | |
| 1.7 | 2.6 | 1.2 | 1.2 | 7.0 | |
| Mean±S.D. | 0.9±0.6 | 1.8±0.7 | 0.7±0.2 | 2.2±0.8¥ | 5.4±1.5# |
| 15 months | |||||
| 0.8 | 1.9 | 0.5 | 1.4 | 3.3 | |
| 1.0 | 2.0 | 0.9 | 2.4 | 6.7 | |
| 1.5 | 1.4 | 1.3 | 2.0 | 4.5 | |
| 0.3 | 1.9 | 0.4 | 1.8 | 5.8 | |
| 0.6 | 0.7 | 0.5 | 0.7 | 7.0 | |
| Mean±S.D. | 0.8±0.5 | 1.5±0.5 | 0.6±0.4 | 1.7±0.7*)¥ | 5.4±1.5# |
A high level of DNA breakage was observed in the brains of 15-month-old rats following SD only. This result suggests that obesity status does not affect genetic damage in brain cells (Table 3. The blood, liver, and brain cells were further assayed with hydrogen peroxide to ensure the sensitivity of the assay. Sensitivity was confirmed by statistically significant differences (p<0.05), which were detected when compared to negative controls on all organs evaluated.
DNA damage (expressed as tail moment data) in the brain tissues of lean and obese Zucker rats that underwent sleep deprivation.
| Age | DNA damage (tail moment) | ||||
|---|---|---|---|---|---|
| Control | Sleep Deprivation | Positive control1 | |||
| Lean | Obese | Lean | Obese | ||
| 3 months | |||||
| 0.6 | 0.8 | 1.9 | 4.4 | 4.5 | |
| 1.2 | 1.2 | 1.2 | 3.7 | 6.2 | |
| 0.6 | 0.6 | 1.0 | 1.6 | 3.6 | |
| 0.5 | 0.1 | 1.5 | 2.4 | 7.2 | |
| 1.4 | 0.2 | 1.7 | 0.8 | 5.5 | |
| Mean±S.D. | 0.8±0.4 | 0.5±0.5 | 1.5±0.3 | 2.6±1.5 | 5.4±1.4# |
| 6 months | |||||
| 1.5 | 2.0 | 2.5 | 3.3 | 4.5 | |
| 1.3 | 1.4 | 2.0 | 2.7 | 6.2 | |
| 0.8 | 0.5 | 1.1 | 1.7 | 3.6 | |
| 0.4 | 0.3 | 2.4 | 1.3 | 7.2 | |
| 0.6 | 0.2 | 0.9 | 1.9 | 5.5 | |
| Mean±S.D. | 0.9±0.5 | 0.8±0.7 | 1.7±0.8 | 2.2±0.7 | 5.4±1.4# |
| 15 months | |||||
| 1.7 | 0.1 | 3.0 | 4.5 | 4.5 | |
| 0.9 | 0.2 | 2.7 | 2.8 | 6.2 | |
| 0.4 | 0.4 | 2.8 | 3.2 | 3.6 | |
| 0.8 | 0.3 | 2.7 | 3.8 | 7.2 | |
| 0.3 | 0.2 | 1.8 | 4.0 | 5.5 | |
| Mean±S.D. | 0.8±0.6 | 0.2±0.1 | 2.6±0.4 | 3.7±0.7*)¥ | 5.4±1.4# |
This study evaluated DNA migration induced by experimental sleep loss in obese versus lean rats. DNA migration was detected by the comet assay in peripheral blood cells from 15-month-old rats.
Based on the tail moment data, the results of this study demonstrated that the comet assay, under our specific experimental conditions, was able to detect the presence of DNA migration in peripheral blood cells from rats at 15 months of age. DNA migration was not detected at younger ages (3 or 6 months), which was possibly because the time allowed was insufficient for a positive genotoxic response to occur on peripheral blood cells. It seems that age plays an important role in the detection of DNA breakage by the comet assay.
The brain was evaluated in this study because it has been shown to be the primary target of DNA damage following SD. In fact, our results demonstrated that extensive genotoxic damage occurred in 15-month-old obese rats subjected to SD. This increase could be partially explained by increased levels of the bigenomic transcripts of oxidative stress (34), which likely contribute to an increase in adenosine triphosphate production during wakefulness (35). Others have also reported that aging impairs the unfolded protein response following SD because of the activation of several pro-apoptotic signaling proteins (36). The results of a study by Kumar et al. suggest that a nitric oxide (NO) mechanism is closely involved in SD-induced behavioral alterations and oxidative damage in mice (37).
In this study, obesity did not lead to DNA migration in brain cells. It is important to note that the comet assay does not necessarily predict the mutagenic potential of all pathological conditions. Moreover, the genotoxicity of SD and obesity status can be modulated in combination with other DNA-damaging agents present in the brain. Altogether, the results from previous studies and this investigation provide evidence that sleep loss is able to induce genetic damage in brain cells, regardless of obesity status or age. In support of these results, Andersen and Ribeiro et al. (16) reported that SD promoted genetic damage in blood and brain cells, particularly following acute exposure.
Chang et al. (38) argue that SD predisposes the liver to oxidative stress and phospholipid damage, leading to injury of the genetic apparatus. Nevertheless, our results revealed that the comet assay failed to detect DNA breakage in liver cells of rats subjected to SD but detected DNA damage in liver cells from obese rats. These findings corroborate data that indicate that genotoxicity in liver cells is associated with obesity in Zucker rats (38).
These findings are biologically relevant because they demonstrate that obesity may negatively impact the incidence, severity, and clinical course of many types of chronic liver diseases and that obesity may be a causative agent for injuries such as nonalcoholic steatohepatitis (39,40). Moreover, the results of an epidemiological study have shown that there is a strong, positive correlation between body mass index and the incidence of several types of cancer (2). Nevertheless, age and SD did not modulate the noxious effects observed in the liver cells from these rats.
The development of genetic damage in target cells depends not only on the initial levels of the induced DNA damage and its repair but also on other contributing factors, including the production of reactive metabolites, their distribution, and their effect on cell proliferation. Moreover, genotoxicity tests detect compounds that induce DNA damage directly or indirectly by various mechanisms. However, no single test is able to detect all genotoxic agents. Thus, for more detailed information on the genotoxic potential of SD on liver cells, a series of tests are necessary.
In conclusion, our results revealed that obesity was associated with genetic damage in liver cells, whereas SD was associated with DNA damage in the peripheral blood and brain cells. The results of this study suggest that there is no synergistic effect of these noxious conditions. Because DNA damage is an important event that is related to genomic instability and to several degenerative diseases, the results of this study are a relevant contribution to the evaluation of the potential health risks associated with sleep loss related to chronic diseases such as obesity.
AUTHOR CONTRIBUTIONSTenorio NM, Tufik S, and Andersen ML were involved in the conception and design of the study. Tenorio NM, Alvarenga TA, and Hirotsu C acquired the data. Tenorio NM, Ribeiro DA, Alvarenga TA, Fracalossi AC, and Carlin V analyzed and interpreted the data. Tenorio NM, Ribeiro DA, Alvarenga TA, Fracalossi AC, Carlin V, Hirotsu C, Tufik S, and Andersen ML were responsible for the final approval of the completed article.
The authors gratefully acknowledge the invaluable assistance of Marilde Costa and Waldemarks Leite. FUNDING: This work was supported by grants from Associação Fundo de Incentivo à Pesquisa (AFIP) and Fundação de Amparo à Pesquisa do Estado de São Paulo (CEPID #98/14303-3 to S.T., #11/12325-6 to T.A.A., #10/50129-1 to C.H., #07/01228-4 to D.A.R., #07/01228-4 to A.C.C.F., and #09/03360-2 to V.C.). M.L.A., D.A.R., C.T.B., and S.T. are recipients of CNPq fellowships.
No potential conflict of interest was reported.