



Bardet-Biedl syndrome is a genetic, multisystem disorder that causes severe visual impairment. This condition is characterized by retinal dystrophy, obesity, digit anomalies, renal disease, and hypogonadism. The purpose of this study was to analyze visual acuity and full-field electroretinogram findings in patients with the Bardet-Biedl syndrome phenotype.
METHODS:The visual acuity of a group of 23 patients (15 males) with ages ranging from 6-36 years (mean = 15.8±6.4; median = 14.7) was assessed. Retinal function was evaluated by full-field electroretinography, and dark-adapted thresholds were assessed.
RESULTS:Visual acuity in the better-seeing eye was 20/40 or better in 5 patients (21.7%), 20/50-20/150 in 13 (56.5%) patients, 20/200-20/400 in 2 (8.7%) patients and worse than 20/400 in one (4.3%) patient. The mean acuity in the better-seeing eye was 0.7±0.6 logMAR (20/100, Snellen equivalent). Scotopic rod and maximal responses were non-detectable in 21 (91.3%) patients, and cone responses were non-detectable in 15 (65.2%) patients. Elevated dark-adapted visual thresholds were observed in all 19 patients who were able to be assessed, with 10 (52.6%) patients having thresholds greater than 30 dB.
CONCLUSIONS:In a relatively young cohort of patients with Bardet-Biedl syndrome, only 21% had 20/40 or better vision. ERG scotopic responses were absent in the majority of cases, with cone responses being observed in less than half of cases. These findings showed the early deleterious effects in retinal function and visual acuity caused by this condition.
Bardet-Biedl Syndrome (BBS), first described by Bardet in 1920 and Biedl in 1922, is a rare autosomal recessive disorder that affects numerous tissues and causes severe visual impairment (1). The estimated prevalence of BBS ranges from 1:125,000 in Europe to 1:18,500 in certain isolated populations in Canada (2,3). The clinical diagnosis of BBS is based on the presence of at least four of five cardinal features: retinal dystrophy, dystrophic extremities (polydactyly, syndactyly, brachydactyly), obesity, hypogenitalism in men only, and renal disease (4,5). BBS is currently considered a ‘ciliopathy,‘ with the underlying defects affecting the basal bodies of ciliated cells, such as the connecting cilium in the outer retina (6). BBS patients variably display all common ciliopathy features: polydactyly, cystic kidneys, retinal degenerations, and situs inversus. To date, mutations in any one of 16 different genes can cause this phenotype (7–11).
Ocular manifestations of BBS include a marked reduction of electroretinogram (ERG) amplitude and notable degree of retinal pigmentary degenerative changes in early childhood (1). Retinal function in BBS patients has been studied by full-field ERG and shows a variable expression of rod-cone or cone-rod dysfunction (1,5,12–16). In BBS carriers, abnormal rod-mediated responses have been detected by full-field ERG, and areas of local dysfunction in the central retina have been observed by multifocal ERG (17,18).
The natural course of BBS is characterized by poor visual prognosis, with central vision declining by 1 line per year on the logMAR chart and peripheral vision deteriorating 0.19 log units per year in dark-adapted thresholds (12). The optic disks and retinal vessels are normal in infancy; disk pallor and attenuated retinal vessels develop with age. Pigmentary changes are observed in the peripheral fundus (19,20). Night blindness usually manifests by eight years of age and progresses to complete blindness by 16-20 years of age (7).
Although different forms of BBS can correlate with specific mutations, demonstrating the genetic heterogeneity in this condition, several studies have suggested that molecular investigations do not show clear phenotype-genotype correlations. Therefore, the diagnosis is still made using clinical data (10,21). Moreover, BBS may be characterized by profound inter- and intrafamilial clinical variability, which may be partially explained by the presence of second-site modifiers (9). Because BBS has a variable clinical presentation, the purpose of this retrospective study was to analyze visual acuity (VA) and full-field ERG findings in patients with the BBS phenotype recruited from a public university hospital in Brazil.
MATERIALS AND METHODSParticipantsParticipants were patients with the Bardet-Biedl syndrome phenotype who were referred to the Electrophysiology of Vision Laboratory at the Department of Ophthalmology of the Federal University of São Paulo, São Paulo, Brazil for assessment from October 1998 to August 2010. During this time frame, 3,282 patients were referred to our laboratory, and the Bardet-Biedl syndrome group comprised 0.8% of these patients. A history of consanguinity was obtained by a personal interview. Non-ocular abnormalities were determined by history and/or available medical records; these cardinal features were not specifically assessed in these patients. This study was approved by the Federal University of São Paulo's Committee of Ethics in Research and followed the tenets of the Declaration of Helsinki. Written informed consent was obtained from each patient or his/her parents or guardians after researchers explained the purpose, procedures, and possible risks and benefits of the study.
Retinal dystrophy type was classified according to the standard clinical criteria, considering history, fundus examination and previous full-field ERG responses (recorded from both eyes with bipolar contact lens electrodes) as rod-cone or cone-rod dystrophy. Inclusion criteria were the presence of at least four of the following cardinal features: retinal dystrophy, obesity, extremity abnormalities, hypogenitalism in males, and renal abnormalities.
ProceduresOphthalmic ExaminationAn ophthalmic examination of both eyes was performed by a retina specialist (SSW), including slit-lamp, refraction, and dilated indirect ophthalmoscopy. Best-corrected visual acuity (BCVA) was measured in each eye using a retro-illuminated Early Treatment Diabetic Retinopathy Study Chart built with Tumble “E” optotypes presented at a distance of 4 m. Ocular motility was assessed using cover/uncover and alternate cover testing for distance and near vision testing, with and without glasses, with emphasis on the diagnosis of strabismus and nystagmus.
Full-Field ElectroretinographyA minimum pupil diameter of 6 mm was obtained after administering a drop of tropicamide 1% with a drop of phenylephrine 10%, and all subjects were dark-adapted for 30 min. Under dim red illumination, a bipolar contact lens electrode (Burian-Allen bipolar electrode, Hansen Ophthalmic Development Lab, Coralville, IA, USA) was placed on the corneal surface. The corneal surface was anesthetized with two drops of tetracaine 1.0%, and a drop of methylcellulose 2% was placed on the inside surface of the contact lens for protection and to ensure good electrical contact. A gold cup ground electrode was applied to the earlobe. All stimuli were presented in a Ganzfeld dome (LKC Technologies Inc., Gaithersburg, MD, USA). Signals were amplified (gain, 910,000; 0.3–500 Hz), digitized, averaged, saved, and displayed by a digital plotter (UTAS E-3000 System, LKC Technologies Inc., Gaithersburg, MD, USA). ERGs were recorded using the standard International Society for Clinical Electrophysiology of Vision (ISCEV) protocol (22). Stimulus and recording details have been described previously (23). The peak-to-peak amplitude (μV) and the implicit time (ms) from each step of the ISCEV standard protocol were determined. The oscillatory potential amplitude was calculated as the sum of each wavelet and automatically analyzed by the UTAS E-3000 system.
Dark-adapted thresholdsDark-adapted thresholds were obtained using the Scotopic Sensitivity Tester (SST-1) (LKC Technologies Inc., Gaithersburg, MD, USA). The SST-1 uses a full-field green LED (λmax = 572 nm) as its stimulus in dark adaptometry mode, with a maximum intensity of -2.15 log cd/m2 at 30 dB. The SST-1 stimulus intensity decreases in 1-dB increments and increases in 5-dB increments, which range from 0 to 30 dB. The SST-1 thresholds were obtained using a two-alternative forced-choice staircase protocol (stimulus vs. non-stimulus, n = 3, decreasing in 1-dB increments). The dark-adapted final threshold was obtained by repeating this protocol three times and averaging the results (24).
Statistical AnalysisData from ERG parameters (peak-to-peak amplitude and implicit time) from each step according to the ISCEV standard protocol were measured and compared with normative data from our own laboratory. A normality test was used to confirm the data distribution. A Spearman's rank correlation coefficient was performed to investigate possible associations between a) BCVA in the better-seeing eye and age, b) BCVA and peak-to-peak amplitude for cone responses, and c) BCVA and peak-to-peak amplitude for 30-Hz flicker responses. A result was considered statistically significant if the p-value was less than 0.05.
RESULTSWe examined 23 patients (15 males) from 22 families with ages ranging from 6-36 years (mean = 15.8±6.4; median = 14.7). A summary of the major clinical characteristics is presented in Table 1. A history of consanguinity was found in 11 participants (47.8%). The patients' self-reported race was white in 14 (61%) patients, mixed white/black in eight patients (35%), and black in one patient (4%). The most frequent presenting visual symptoms were as follows: night vision loss in 22 patients (95.6%), visual acuity loss in 19 patients (82.6%), and visual field loss in 13 patients (56.5%). A 12-year-old male patient had no visual symptoms (case #8).
Clinical characteristics of 23 patients with Bardet-Biedl Syndrome.
| Case# | Age(yrs.) | Gender | Race | History of Consanguinity | Presenting Symptoms | BCVA(better-seeing eye) | Refraction(Mean SE) | Ophthalmoscopic Changes | RetinalDystrophy | Nystagmus | Strabismus |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6 | M | mixed | N | VA, NV loss | Uncooperative | NA | Uncooperative | rod-cone | Y | Ortho |
| 2 | 8 | M | white | N | VA, NV loss | 20/100 | +7.00 | MA | rod-cone | N | Ortho |
| 3 | 9 | M | mixed | N | VA, NV loss | 20/40 | +11.75 | AV, DP, WRPEA | cone-rod | N | Ortho |
| 4 | 10 | M | white | Y | VA, NV loss | 20/160 | -11.50 | WRPEA | rod-cone | Y | XT |
| 5 | 11 | M | white | N | VA, NV loss | 20/40 | +4.25 | WRPEA | rod-cone | N | Ortho |
| 6 | 11 | M | white | Y | NV, VF loss | 20/125 | +2.25 | AV, DP, WRPEA | rod-cone | N | Ortho |
| 7 | 12 | F | mixed | N | VA, NV, VF loss | 20/125 | +0.50 | WRPEA | rod-cone | N | X(T) |
| 8 | 12 | M | white | N | None | 20/40 | NA | AV, DP, MA | cone-rod | N | Ortho |
| 9* | 12 | M | white | Y | VA, NV, VF loss | 20/40 | -2.50 | AV, WRPEA | rod-cone | N | ET |
| 10 | 13 | F | white | N | NV, VF loss | 20/125 | -0.50 | MA, PP | rod-cone | Y | XT |
| 11 | 14 | M | mixed | Y | VA, NV, VF loss | 20/160 | +4.50 | AV, MA, PP, WRPEA | cone-rod | Y | Ortho |
| 12 | 14 | M | white | Y | VA, NV, VF loss | 20/100 | +2.00 | MA, WRPEA | rod-cone | N | Ortho |
| 13* | 14 | M | white | Y | VA, NV, VF loss | 20/50 | -2.00 | AV, MA | rod-cone | N | ET |
| 14 | 15 | F | white | N | VA, NV, VF loss | 20/80 | -8.50 | AV, MA | rod-cone | N | Ortho |
| 15 | 15 | F | mixed | N | VA, NV loss | 20/125 | NA | WRPEA | rod-cone | N | Ortho |
| 16 | 16 | F | white | Y | VA, NV loss | 20/80 | -9.25 | AV, DP, WRPEA | rod-cone | N | Ortho |
| 17 | 17 | M | black | Y | VA, NV, VF loss | 20/63 | +1.50 | AV, DP, WRPEA | rod-cone | N | Ortho |
| 18 | 17 | F | white | Y | VA, NV, VF loss | 20/125 | -4.75 | AV, DP, MA, WRPEA | rod-cone | N | Ortho |
| 19 | 18 | M | white | N | VA, NV, VF loss | 20/50 | NA | AV, DP, MA, PP, WRPEA | rod-cone | N | Ortho |
| 20 | 19 | M | mixed | Y | NV, VF loss | 20/25 | +2.25 | AV, DP, PP | rod-cone | N | Ortho |
| 21 | 20 | F | white | Y | VA, NV loss | 20/200 | NA | AV, WRPEA | rod-cone | N | Ortho |
| 22 | 28 | M | mixed | N | VA, NV loss | Uncooperative | NA | AV, DP, PP | rod-cone | Y | ET |
| 23 | 36 | F | mixed | N | VA, NV, VF loss | LP | -6.50 | NA | rod-cone | Y | Ortho |
brothers from a consanguineous marriage; mixed — black/white; BCVA – best-corrected visual acuity; SE – spherical equivalent; VA – visual acuity; NV – night vision; VF – visual field; NA – not available; MA – macular abnormalities; AV – attenuated vessels; DP – disk pallor; WRPEA – widespread retinal pigmented epithelium abnormalities; PP – peripheral pigmentation; ET – esotropia; XT – exotropia; and X(T) – intermittent exotropia.
High ametropias were found in six patients (26.0%), with four patients reporting high myopia (SE≤-6.0 D) and two reporting high hyperopia (SE≥6.0 D). Severe retinal dystrophy was found in all patients. A rod-cone pattern was observed in 20 patients (86.9%), with the remaining three patients showing cone-rod dystrophy. Ophthalmoscopic changes were classified according to fundus photographs (when available) or fundus descriptions. Data were available from 21 of the 23 examined patients. The most frequent fundus changes were attenuated vessels in 14 patients (67.0%), widespread retinal epithelium abnormalities in 14 patients (67.0%), disk pallor in nine patients (42.8%), macular abnormalities in nine patients (42.8%), and peripheral pigmentation in five patients (23.8%).
Six patients (26.0%) presented manifest bilateral nystagmus. On the cover test, three subjects presented esotropia, two presented exotropia, and one presented intermittent exotropia; the remaining 17 subjects had orthoposition of the visual axes. Both nystagmus and strabismus were found in three patients (13.0%).
Visual AcuityThe BCVA distribution from 21 BBS patients is shown in Table 1. BCVA in the better-seeing eye was 20/40 or better in five patients (21.7%), 20/50-20/150 in 13 patients (56.5%), 20/200-20/400 in two patients (8.7%), and worse than 20/400 in one patient (4.3%). Two patients were uncooperative in the visual acuity measurement. The mean VA was 0.7±0.6 logMAR (20/100, Snellen equivalent) in the better-seeing eye and 0.9±0.6 logMAR (20/160, Snellen equivalent) in the worse-seeing eye. No significant correlation was found between age and BCVA (logMAR) in the better-seeing eye (p = 0.24).
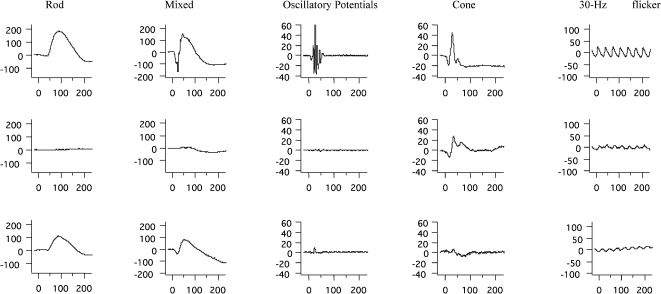
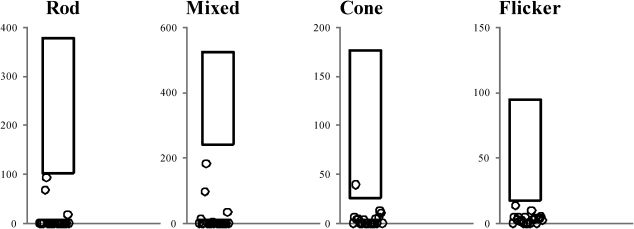
Full-field ERGERG abnormalities were found in all 23 patients. Representative standard full-field ERG recordings from one eye from a healthy normal subject, one patient with rod-cone dystrophy (case #2), and one patient with cone-rod dystrophy (case #3) are shown in Figure 1. Figure 2 summarizes the ERG parameters for scotopic and photopic retinal function from BBS patients compared with our own normative data (mean±2 SD), as represented by rectangles, similar to the previously shown graphic model (13,25). Rod scotopic responses were non-detectable in 20 patients, with only three patients reporting remaining rod function below the lower normal limit. Four patients had detectable mixed scotopic responses, with two patients responding (cases #8 and #3) within the normal limits and two patients (cases #9 and #16) responding below the lower normal limits; the remaining patients had non-detectable responses. Cone function assessed by single-flash cone responses was present in nine patients; all patients except case #2 had responses below the normal limits. Cone responses for 30-Hz flicker were detected in 16 patients and non-detectable in seven patients; all detected responses were below the normal limits. No correlation was found between BCVA and peak-to-peak amplitude for cone responses (p = 0.08) or between BCVA and peak-to-peak amplitude for 30-Hz flicker responses (p = 0.76).
 with amplitudes in μV (Y axis) and time in ms (X axis) from a healthy age-matched subject (upper panels), a patient with Bardet-Biedl syndrome (case #2) presenting rod-cone dystrophy (middle panels), and a patient with Bardet-Biedl syndrome (case #3) presenting cone-rod dystrophy (lower panels).")
Representative standard full-field ERG waveforms (rod, mixed, oscillatory potentials, cone, and 30 Hz-flicker responses) with amplitudes in μV (Y axis) and time in ms (X axis) from a healthy age-matched subject (upper panels), a patient with Bardet-Biedl syndrome (case #2) presenting rod-cone dystrophy (middle panels), and a patient with Bardet-Biedl syndrome (case #3) presenting cone-rod dystrophy (lower panels).
.")
Elevated dark-adapted visual thresholds were found in all 19 patients who were able to be assessed, with 10 patients (52.6%) having thresholds higher than 30 dB. The mean threshold was 22.7±9.04 dB with a median of 30 dB.
Non-ocular findingsOf the 23 patients, 21 patients (91.3%) were obese, 18 patients (78.3%) presented digital anomalies, 18 patients (78.3%) had mental retardation, and 7 patients (30.4%) had renal anomalies.
DISCUSSIONThe present study showed no correlation between BCVA in the better-seeing eye and age, thereby corroborating previous reports in cohorts of three genetic BBS variants (1,16). However, other reports have shown decreased vision in the first decades of life, as found in our studied sample (5,21). Good BCVA in the better-seeing eye (≥20/40) was found in only five patients (21.7%) in our study. These results are similar to those results reported in a group of 10 BBS patients, with four patients showing good visual acuity (16). Legal blindness (BCVA in the better-seeing eye ≤20/200) was present in two (8.7%) older patients (cases #21 and 23). Longitudinal data from these patients should be pursued to detect the visual acuity loss rate (in lines/year), as previously demonstrated (12). The frequency of nystagmus or strabismus was 26%. In six patients with strabismus, there was a fair distribution between esotropias and exotropias. Similar findings have been described in young BBS patients presenting nystagmus, but a tendency to exotropia was not found in our cohort (20,21).
The lack of correlations between BCVA in the better-seeing eye and peak-to-peak amplitude for cone responses and between BCVA and peak-to-peak amplitude for 30-Hz flicker responses was not unusual. Because full-field ERG may not be sensitive enough to detect minimal regional responses from the central retina, these findings confirm previous reports (16,19).
The rod-cone degeneration with no rod responses and elevated dark-adapted thresholds observed in the vast majority of patients confirm previous reports (13,15). Measurable cone-flicker responses were detected in 28/36 patients, and these responses can be used to objectively follow the natural history of the disease (13). In the current study, all patients had severely reduced or extinguished ERG parameters. These findings were also seen in previous studies in young patients and a series of patients with BBS mutations, with younger patients showing less photoreceptor degeneration (1,12). These data show that BBS retinal disease, which is primarily a degeneration of photoreceptors, must affect functional processes common to both rods and cones. Both central (acuities) and more peripheral (thresholds) functions are affected (12). Beside functional abnormalities measured by ERG, disrupted inner and outer segment layers and thinned RPE have been described in vivo by Fourier-domain OCT with inner retinal layer preservation (6,16).
Male predominance was found in our cohort, with a male-to-female ratio of 15∶8 being observed. These results are consistent with previously reported findings in both adults and pediatric BBS patients (20,21). However, other studies have described a slight female predominance (5). These discrepancies could be explained by the low incidence of this recessive condition, which leads to studies with a limited number of participants.
In most developing countries, access to modern molecular diagnostic techniques is extremely limited, reinforcing the need for a detailed phenotypic description to achieve precise clinical diagnoses of several genetic eye disorders, including BBS. In this highly variable disease, not all features are present, as described in a recent case report of two brothers with proven BBS1 mutation without obesity (26). Efforts to make these molecular diagnostic techniques available to all patients with genetic ocular diseases should be pursued.
ERG scotopic responses were absent in the vast majority of cases, and cone responses remained in less than half of patients. These results confirm the early deleterious effects in retinal function and visual acuity caused by BBS.
AUTHOR CONTRIBUTIONSBerezovsky A designed, conducted and analyzed the material; prepared, reviewed and approved the manuscript. Salomão SR designed the study and analyzed the material; prepared, reviewed and approved the manuscript. Rocha DM collected and analyzed data; reviewed and approved the manuscript. Sacai PY collected data; reviewed and approved the manuscript. Watanabe SS collected and analyzed data; reviewed and approved the manuscript. Cavascan NN collected data, reviewed and approved the manuscript.
This study was supported by 1) Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) grants #04/02669-6 and 05/56459-5 to AB; 2) Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brasília, Brazil grant #474251/2009-8 to AB; and 3) Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília, Brazil) research scholarships to AB and SRS.
No potential conflict of interest was reported.