




La región selar y paraselar es una área anatómica compleja en la que se pueden desarrollar una serie de enfermedades. La glándula hipofisaria puede verse afectada por una amplia gama de trastornos, que cursan con características clínicas similares. El diagnóstico de estas lesiones implica un enfoque multidisciplinar y, junto con la exploración clínica, analítica, radiológica y quirúrgica, el estudio histológico de los adenomas hipofisarios determina la conducta que tomará el médico especialista ante el paciente. Con la aparición, en los últimos años, de nuevas técnicas inmunohistoquímicas, la clasificación histopatológica se ha vuelto más compleja y amplia, ya que además de ser el gold standard del diagnóstico, tiene implicaciones pronósticas. El objetivo de esta revisión es actualizar conceptos del diagnóstico histológico de la patología hipofisaria más frecuente, de manera clara y fácil, especialmente para aquellos profesionales en contacto directo con este tipo de patología.
The sellar and parasellar region is a complex anatomical area in which several diseases may develop. The pituitary gland may be affected by a wide range of conditions having similar clinical characteristics. Diagnosis of these lesions requires a multidisciplinary approach including, in addition to clinical, laboratory, imaging, and surgical findings, histological diagnosis of pituitary adenomas to guide therapeutic management. As the result of development in recent years of new immunohistochemical techniques, histopathological classification has become more complex and wide, and not only continues to be the gold standard in diagnosis, but also has prognostic implications. The aim of this review is to provide a clear and simple update of the main concepts of histological diagnosis of the most common pituitary conditions, especially for professionals in direct contact with such diseases.
El abordaje de la patología hipofisaria y región selar es complejo, ya que numerosos tumores y lesiones seudotumorales pueden afectar esta área, lo que requiere el conocimiento de muchas entidades patológicas. Los tumores de la glándula hipofisaria y región selar representan aproximadamente el 15% de todos los tumores cerebrales1; en su gran mayoría corresponden a adenomas hipofisarios (AH) (85%), seguidos de craneofaringiomas (3%), quistes de la hendidura de Rathke (2%), meningiomas (1%) y metástasis (0,5%); el resto, son lesiones más raras2, aunque mimetizan al AH en los estudios de neuroimagen, por lo que el diagnóstico definitivo recae sobre el patólogo.
Como resultado del desarrollo y amplio uso de los estudios de imagen neurorradiológicos, tomografía computarizada y resonancia magnética, cada vez se diagnostican con más frecuencia lesiones hipofisarias clínicamente silentes3–5. En la actualidad, la exploración por resonancia magnética se considera la modalidad preferida para el diagnóstico de las lesiones hipofisarias, debido a su capacidad de examinar múltiples planos y la posibilidad de diferenciar los tejidos blandos en función de su captación de contraste. Una hipointensidad focal dentro de la hipófisis se considera anómala y sugiere un adenoma.
Numerosos tipos de lesiones, seudotumorales y tumorales, pueden afectar la hipófisis y la región selar (anomalías del desarrollo, quistes, enfermedades inflamatorias, infecciosas, metabólicas, neoplásicas y trastornos vasculares), reflejando la compleja anatomía de esta área. En esta revisión nos centraremos en el diagnóstico histológico de la patología hipofisaria más frecuente y relevante.
Tumores de la adenohipófisisCaracterísticas generales de los adenomas hipofisariosLos AH incidentales pueden encontrarse en cerca del 10% de las autopsias6–8. En una reciente revisión de estudios de autopsia y RM, la prevalencia global estimada de AH fue del 16,7%9. Comparativamente, los tumores primarios de la neurohipófisis son más raros y, en general, son similares a los tumores primarios del sistema nervioso central. Sin embargo, la neurohipófisis es un sitio común para las metástasis10.
Los AH son tumores epiteliales benignos derivados de las células intrínsecas de la adenohipófisis. Afectan a ambos sexos, predominantemente entre la 3.ª y 6.ª década11, pudiendo afectar a cualquier grupo etario1,12. Los AH infantiles son extremadamente raros, no obstante cuando se producen por lo general son adenomas secretores de ACTH13. Los AH no son homogéneos; cada subtipo tiene su propia presentación clínica, tendencia para la invasión, patrón de secreción hormonal, características histopatológicas y tratamiento. Los mecanismos implicados en la génesis y progresión tumoral todavía no se conocen bien.
Clínicamente, se clasifican en dos grupos: funcionantes y no funcionantes, dependiendo de que exista o no un síndrome endocrino específico. Alrededor de un tercio de los AH no se asocian con ninguna evidencia clínica o bioquímica de exceso hormonal14; son adenomas clínicamente no funcionantes, que se suelen presentar con signos y síntomas relacionados con el efecto de masa local como dolor de cabeza, déficits neurológicos de los nervios craneales (incluyendo alteraciones del campo visual) e hiperprolactinemia. Esta es debida a la compresión del tallo hipofisario (el denominado «stalk effect»), que impide la llegada de dopamina a la adenohipófisis (y no debe ser malinterpretada por el patólogo como un adenoma productor de prolactina).
En base a su tamaño y características anatómicas, se dividen en microadenomas (<1cm de diámetro), macroadenomas (>1cm y <4cm) y adenomas gigantes (>4cm). Radiológicamente, se han propuesto varias clasificaciones para valorar su extensión e invasividad local, siendo la de Hardy y la de Knosp unas de las más utilizadas15,16.
Los AH también se clasifican histopatológicamente de acuerdo con el contenido hormonal de las células tumorales demostrado por estudio inmunohistoquímico (IHQ) que proporciona información muy relevante para la práctica clínica17. En este artículo seguiremos el esquema clasificatorio para los tumores de la glándula hipofisaria publicado por la Organización Mundial de la Salud (OMS) en 200418.
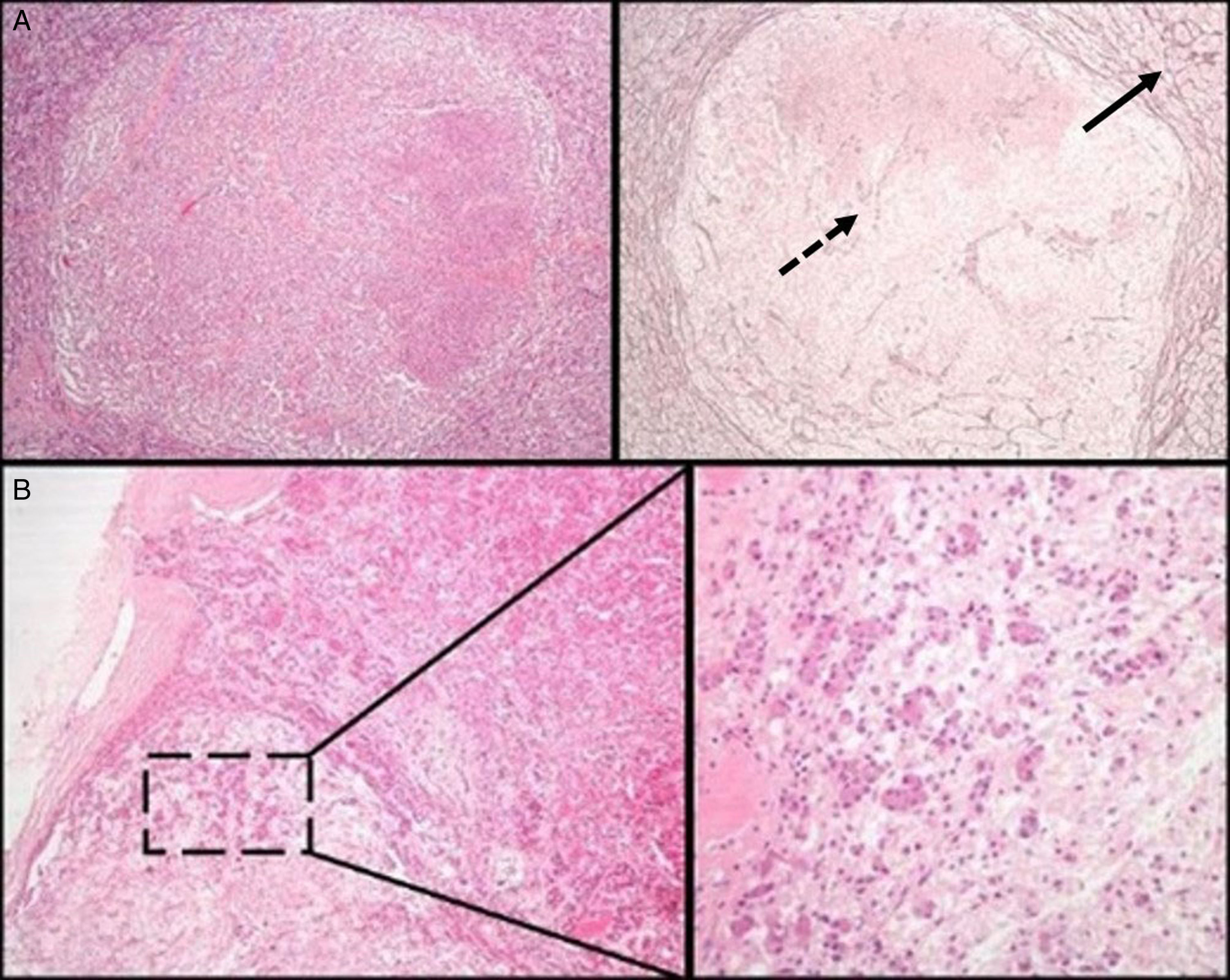
Evaluación patológica inicial de una lesión hipofisariaLa primera decisión que debe tomarse ante un espécimen quirúrgico de esta glándula, es si el tejido sometido para análisis es hipófisis normal o un AH. Para esto, después de la hematoxilina-eosina (HE) la coloración histoquímica más valiosa es la técnica de reticulina, que ayuda a distinguir el patrón acinar conservado de la adenohipófisis normal, de la disrupción de la red de reticulina observada en el AH19 (fig. 1A). La HE y otras técnicas histoquímicas especiales como el periodic acid-Schiff (PAS)-orange G (hoy obsoleta y ampliamente sustituida por la técnica IHQ), ayudarán a visualizar la variedad de tipos de células existentes con diferentes capacidades tintoriales citoplasmáticas (acidófilas, basófilas o cromófobas) de la adenohipófisis normal. En el estudio IHQ, tanto la adenohipófisis normal como el AH son inmunorreactivos para sinaptofisina (un marcador de tumores neuroendocrinos); la positividad para hormonas hipofisarias específicas pone de manifiesto la gran variedad celular observada en los fragmentos de glándula hipofisaria anterior normal (al contrario de lo que sucede en la mayoría de AH). En ocasiones, se pueden encontrar pequeños fragmentos de neurohipófisis normal, especialmente si el neurocirujano ha resecado una lesión quística de la hendidura de Rathke. La mejor técnica IHQ para confirmar la presencia de glándula hipofisaria posterior es la marcación con neurofilamentos, que ayuda también a distinguirla de otras lesiones como el pituicitoma. La escisión de pequeños fragmentos de neurohipófisis no suele tener consecuencias clínicas permanentes aunque sí una diabetes insípida transitoria que normalmente se resuelve después de algunos días. Conviene recordar una variante de la normalidad en la hipófisis posterior que no debe ser confundida con infiltración tumoral, la denominada «invasión basófila» propia del envejecimiento, constituida por pituicitos normales de la hipófisis anterior inmunorreactivos a ACTH que se extienden a la neurohipófisis8 (fig. 1B).
 Hipófisis normal versus adenoma hipofisario. Nótese el patrón acinar periférico de la glándula hipofisaria anterior normal (flecha continua), en contraste con la disrupción de la red de reticulina habitual en un adenoma (flecha discontinua) (técnica histoquímica de HE –izquierda– y de Gomori-reticulina –derecha–, 40×). B) La glándula hipofisaria normal manifiesta una «invasión basófila» fisiológica durante el envejecimiento. Se observan regueros de células endocrinas basófilas que se extienden desde la interfase del lóbulo anterior hasta la neurohipófisis (HE 40×; HE 200x).")
A) Hipófisis normal versus adenoma hipofisario. Nótese el patrón acinar periférico de la glándula hipofisaria anterior normal (flecha continua), en contraste con la disrupción de la red de reticulina habitual en un adenoma (flecha discontinua) (técnica histoquímica de HE –izquierda– y de Gomori-reticulina –derecha–, 40×). B) La glándula hipofisaria normal manifiesta una «invasión basófila» fisiológica durante el envejecimiento. Se observan regueros de células endocrinas basófilas que se extienden desde la interfase del lóbulo anterior hasta la neurohipófisis (HE 40×; HE 200x).
La segunda decisión a tomar será si la lesión es o no un AH. La mayor parte de estos tumores se presentan con un patrón de crecimiento difuso; sin embargo, puede haber variaciones ocasionales en su arquitectura (patrón sinusoidal, macronodular o festoneado) que no tienen relación con el pronóstico, pero que pueden confundir a la hora de hacer el diagnóstico. Otras características que podemos encontrar son células de citoplasma claro, quistes de tamaños variados, hendiduras producidas por cristales de colesterol, macrófagos xantomatosos e incluso procesos adaptativos como la metaplasia ósea (que debe distinguirse de la invasión ósea del suelo de la silla turca por el adenoma, que no suele causar reacción osteoblástica y en la cual las trabéculas óseas se hacen más finas)19.
En cuanto a las hormonas adenohipofisarias específicas necesarias para la subtipificación de los AH, recomendamos anticuerpos contra PRL, GH, ACTH, FSH, LH y TSH como panel IHQ mínimo. A estos se añadirán marcadores de pronóstico, concretamente el marcador de proliferación celular Ki-67 y el marcador del gen supresor tumoral p53, para diagnóstico diferencial entre AH típicos y atípicos. Debido a la dificultad que a veces supone la distinción entre núcleos apoptóticos y mitosis, es recomendable también la utilización del anticuerpo fosfohistona H3 (PHH3); una vez que la histona H3 (una proteína del núcleo de la histona constituyente proteínico principal de la cromatina) no es fosforilada durante la apoptosis20, puede servir para separar las figuras mitóticas de los cuerpos apoptóticos y detritus cariorrécticos.
El uso IHQ de la vimentina, proteína ácida fibrilar glial o la proteína S-100, no tienen valor en el diagnóstico y subtipificación de AH, y no se recomienda su uso en la IHQ básica inicial, aunque pueden ser usados cuando las características del tumor al microscopio óptico sugieren una lesión de células fusiformes de la región selar.
Adenomas secretores de prolactinaLos adenomas secretores de prolactina, también llamados prolactinomas, representan cerca del 80% de los adenomas funcionantes y aproximadamente el 40-50% de todos los AH21,22, siendo el tipo más frecuente de AH23; sin embargo, la prevalencia de prolactinomas en series quirúrgicas tiende a ser baja, dada su buena respuesta al tratamiento médico (la mayoría de los pacientes con este tipo de tumores son tratados en primera línea con agonistas de la dopamina). El diagnóstico se confirma por hiperprolactinemia sostenida y la evidencia neurorradiológica de un tumor hipofisario, descartando otras causas de hiperprolactinemia. Histológicamente, la IHQ muestra reactividad para PRL con un patrón de marcación perinuclear característico en «dot», conocido como patrón de Golgi. Los agonistas de la dopamina son fármacos que actúan directamente sobre las células tumorales induciendo atrofia y la consecuente reducción tumoral. En estos casos, a nivel histológico podemos encontrar células tumorales más pequeñas, con reducción del citoplasma y núcleos hipercromáticos, además de varios grados de fibrosis tumoral perivascular e intersticial. La identificación IHQ de prolactina en el adenoma, apoyaría la terapia postoperatoria con agonistas de la dopamina si persiste tumor residual o hipersecreción. En el análisis ultraestructural, los prolactinomas pueden dividirse en densamente y escasamente granulados, aunque la importancia clínica de esta distinción es cuestionable24.
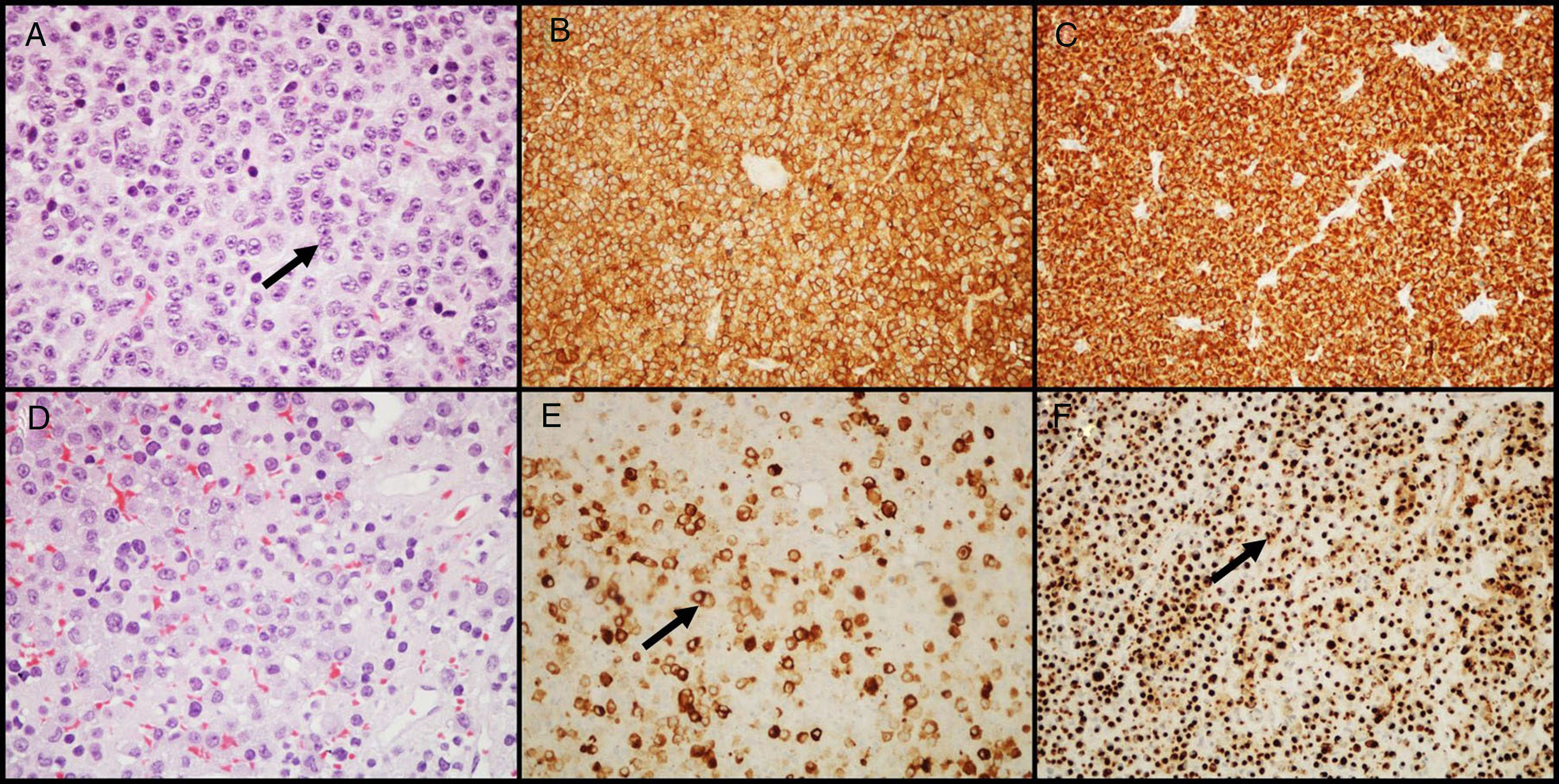
Adenomas secretores de GHLos adenomas secretores de GH representan cerca de 20% de los AH. Los pacientes se presentan con signos y síntomas de gigantismo o acromegalia, así como niveles altos en suero de GH e IGF-I11. En aproximadamente el 30-50% de los pacientes hay cosecreción tumoral de prolactina, lo que resulta en signos y síntomas de hiperprolactinemia. Histológicamente, la cantidad de gránulos secretores presentes en el citoplasma celular caracteriza dos tipos de adenomas: densamente granulados (caracterizados por un citoplasma granular eosinófilo de las células tumorales) y escasamente granulados (compuesto por células tumorales más pequeñas, con citoplasma cromófobo y núcleo excéntrico). En los primeros, la IHQ muestra marcación intensa y difusa para GH y el factor de transcripción Pit-1; en los segundos, la marcación para GH es heterogénea y menos intensa, además de que en el citoplasma pueden observarse estructuras eosinófilas paranucleares llamadas «cuerpos fibrosos»25 (una acumulación de filamentos intermedios y retículo endoplásmico), mejor visualizadas con IHQ para citoqueratinas 8/18 (fig. 2). Otro anticuerpo que puede ser de utilidad para distinguir ambos subtipos, es la E-caderina, ya que hay pérdida de expresión en los adenomas secretores de GH escasamente granulados, pero no en los densamente granulados26. La distinción entre estos 2 subtipos de adenomas es importante, una vez que tienen diferente comportamiento clínico (los escasamente granulados exhiben un comportamiento biológico más agresivo y responden menos al tratamiento con ligandos de receptores de la somatostatina)27,28. Gran número de adenomas secretores de GH pueden mostrar inmunorreactividad secundaria para otras hormonas hipofisarias (PRL, FSH, LH o β-TSH)27,29. En aquellos adenomas tratados con ligandos de receptores de la somatostatina, principalmente octreótido, los cambios más comunes son diversos grados de fibrosis perivascular e intersticial.
 Los adenomas que secretan GH densamente granulados muestran células grandes con un citoplasma granular eosinófilo y un núcleo central con nucléolos prominentes (flecha); B) el tumor muestra una inmunomarcación intensa y difusa para GH; C) la inmunomarcación con citoqueratina muestra reactividad difusa citoplasmática. D) Los adenomas que secretan GH escasamente granulados son característicamente más cromófobos que los densamente granulados; E) la marcación de GH es heterogénea y menos prominente (flecha); F) la inmunohistoquímica con citoqueratina destaca los cuerpos fibrosos (flecha). (A y D - HE 400×; B y E - GH 200×; C y F - citoqueratinas 8/18 200×).")
Adenomas secretores de GH. A) Los adenomas que secretan GH densamente granulados muestran células grandes con un citoplasma granular eosinófilo y un núcleo central con nucléolos prominentes (flecha); B) el tumor muestra una inmunomarcación intensa y difusa para GH; C) la inmunomarcación con citoqueratina muestra reactividad difusa citoplasmática. D) Los adenomas que secretan GH escasamente granulados son característicamente más cromófobos que los densamente granulados; E) la marcación de GH es heterogénea y menos prominente (flecha); F) la inmunohistoquímica con citoqueratina destaca los cuerpos fibrosos (flecha). (A y D - HE 400×; B y E - GH 200×; C y F - citoqueratinas 8/18 200×).
Los adenomas mixtos secretores de GH y PRL constituyen aproximadamente el 8% de los AH2. Los pacientes con este tipo de tumores presentan signos y síntomas de ambos, acromegalia e hiperprolactinemia27. El diagnóstico de este grupo de adenomas requiere un análisis más complejo, IHQ y ultraestructural, siendo su distinción fundamental, ya que tiene implicaciones clínicas y pronósticas. Morfológicamente se pueden identificar tres subtipos: 1) adenomas mixtos de células secretoras de GH y de células secretoras de PRL; 2) adenomas de células somatotropas y 3) adenomas de células madre acidófilas27,30. En el primer subtipo, la IHQ demuestra marcación para GH y PRL con diversos grados de intensidad y distribución. A nivel ultraestructural se observan dos poblaciones celulares separadas. Los adenomas de células somatotropas son tumores raros (menos del 2% de todos los AH y cerca de 8% de los tumores asociados a acromegalia31,32) en los que la IHQ demuestra marcación para ambos, GH y PRL, en el mismo tipo de célula tumoral. El análisis ultraestructural demuestra un adenoma bien diferenciado compuesto por una población de células monomorfas que contienen características de células secretoras de GH y de PRL. Los adenomas de células madre acidófilas son muy raros27 y su diagnóstico tiene gran importancia clínica, ya que pueden confundirse con prolactinomas una vez que la mayoría de los pacientes presentan características de hiperprolactinemia33. Histológicamente son tumores cromófobos, con cambios oncocíticos citoplasmáticos focales. El estudio IHQ muestra marcación para PRL, y en menor medida para GH, en el citoplasma de las mismas células tumorales. Es necesaria microscopía electrónica para la identificación precisa de estos adenomas33, donde se pueden observar megamitocondrias, responsables de la apariencia oncocítica en microscopía óptica.
Adenomas secretores de ACTHLos adenomas secretores de ACTH asociados con enfermedad de Cushing representan aproximadamente 10-15% de todos los AH34. Histológicamente son comunes las formaciones papilares, y a menudo se puede ver una intensa marcación con la técnica histoquímica PAS y con la IHQ para ACTH. Ocasionalmente se pueden observar en el citoplasma haces hialinos periféricos, lo que provoca apariencia de «células en diana»; estos cambios se denominan hialina de Crooke y corresponden a la acumulación de filamentos intermedios de citoqueratina (la IHQ para citoqueratina muestra esta acumulación intracitoplasmática); parece ser un efecto directo de los altos niveles séricos de cortisol sobre estas células hipofisarias35.
Adenomas secretores de gonadotrofinasLos adenomas secretores de gonadotrofina (adenomas que secretan FSH y LH) constituyen alrededor del 20% de todos los AH36. Son adenomas que generalmente no causan un síndrome clínico relacionado con la sobreproducción hormonal y clínicamente pasan por ser adenomas no funcionantes. Histológicamente las células tumorales suelen estar dispuestas en un patrón de crecimiento difuso, pero es frecuente la formación de estructuras papilares alrededor de vasos sanguíneos36, dando lugar a un patrón que asemeja la formación de seudorrosetas perivasculares. Para su caracterización IHQ se recomienda la utilización de anticuerpos monoclonales específicos contra β-FSH (la más frecuente36), β-LH y alfa-subunidad (α-SU), debido a que estas lesiones pueden mostrar diversos grados de reactividad para una o más subunidades de gonadotrofina. La caracterización de estos adenomas por microscopía electrónica puede ser de interés científico, pero no altera el manejo clínico de estos pacientes.
Adenomas secretores de TSHLos adenomas secretores de TSH son los AH menos frecuentes37 (menos del 1% de todos los adenomas). La IHQ suele revelar positividad variable para β-TSH y comúnmente también para α-SU. El diagnóstico puede ser problemático si la presentación clínica y la inmunorreactividad para TSH no son convincentes. En estos casos la microscopía electrónica es obligatoria para un diagnóstico adecuado.
Adenomas silentesHay algunos AH clínicamente no funcionantes, en los que a pesar de faltar el síndrome clínico o el signo de hipo- o hipersecreción hormonal, presentan un patrón de marcación IHQ y un aspecto ultraestructural consistentes con un adenoma secretor. Son los denominados adenomas silentes. De estos, los que tienen implicaciones clínicas más significativas son los adenomas corticotropos «silenciosos», que se caracterizan por inmunorreactividad para ACTH (con ausencia de cualquier signo clínico de enfermedad de Cushing o niveles séricos que reflejen exceso de secreción de ACTH). Característicamente, este tipo de adenomas muestran una alta tendencia para la hemorragia y la apoplejía (definida como la aparición repentina de síntomas tales como dolor de cabeza intenso, náuseas, vómitos, pérdida de visión, parálisis de nervios craneales, y alteración de la conciencia con evidencia radiológica de infarto hemorrágico del AH, seguido con frecuencia de hipopituitarismo38), presentándose en cerca de un tercio de los pacientes39,40.
Adenomas plurihormonalesLos adenomas plurihormonales son adenomas raros, que tienen inmunorreactividad inusual para múltiples hormonas hipofisarias que no están relacionadas a través de la citogénesis ni del desarrollo normal de la hipófisis anterior41.
Adenomas de células nulasAproximadamente el 20% de los AH no muestran evidencia clínica ni IHQ de producción hormonal14,42. A estos tumores se les denomina adenomas de células nulas, basado en gran parte en la ausencia de características ultraestructurales que proporcionan diferenciación específica. Histológicamente, algunos de estos casos pueden mostrar cambios oncocíticos celulares y, debido a esto, la designación de oncocitoma podrá ser aplicada a esos adenomas42. Existe una considerable sobreposición entre los adenomas de células nulas y los adenomas gonadotropos, una vez que se ha encontrado que algunos de estos adenomas muestran inmunorreactividad débil y focal para hormonas glucoproteicas. No obstante, desde el punto de vista del manejo del paciente, la diferenciación entre estos dos adenomas tiene poca importancia clínica14.
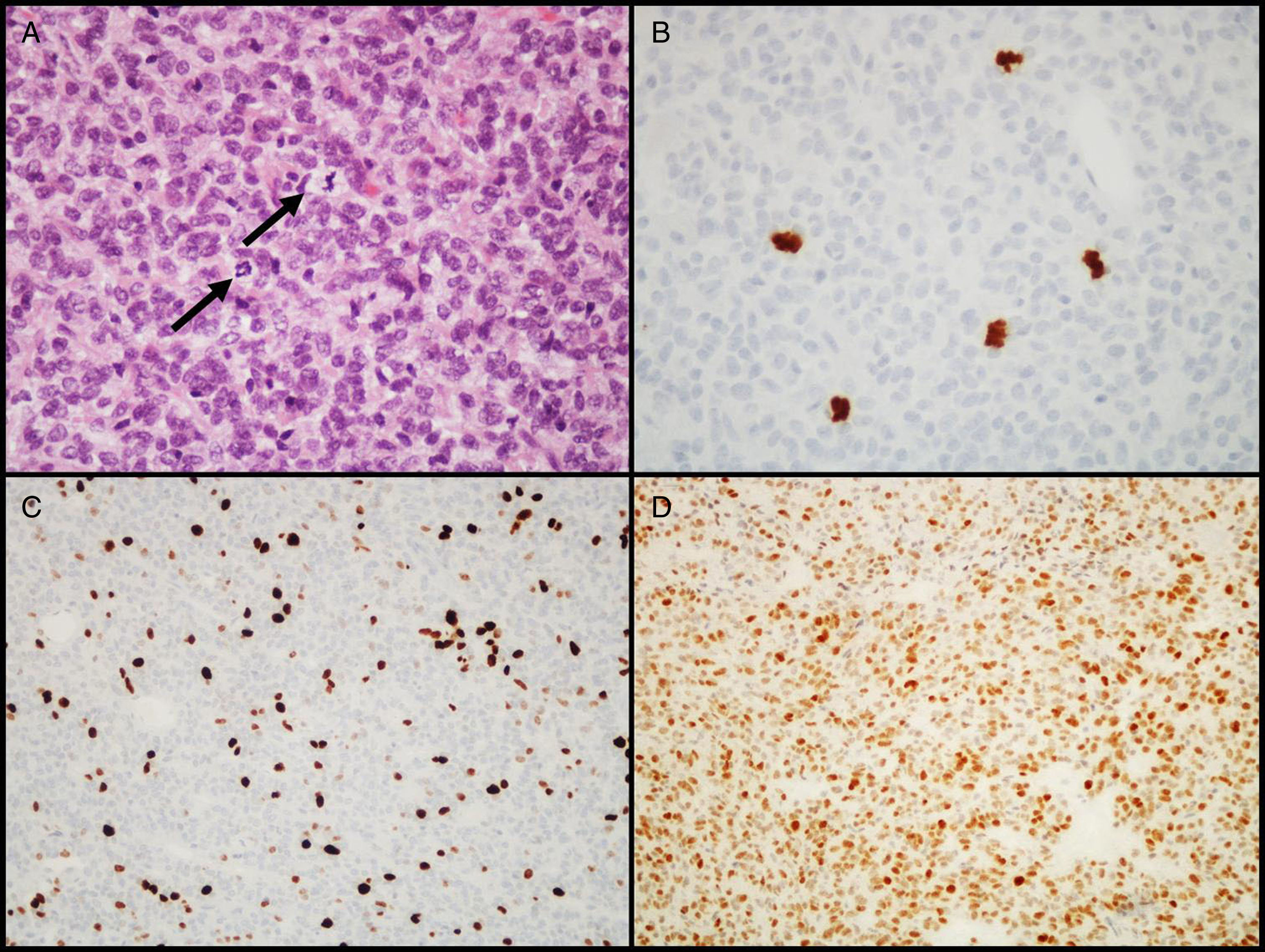
Adenomas atípicosEn 2004, la OMS introdujo la designación de adenoma atípico para aquellos tumores que muestran características histológicas sugestivas de comportamiento clínico agresivo (como crecimiento invasivo)18. Estos tumores se caracterizan por tener un índice mitótico elevado, un índice de proliferación celular (Ki-67) superior a 3% y extensa inmunopositividad para la proteína p5318 (fig. 3). Esta última clasificación de la OMS generó alguna controversia, ya que las diferencias entre AH «típicos» y «atípicos» no están claramente definidas, al no establecer valores de corte para criterios como el número de mitosis, o el porcentaje de núcleos positivos e intensidad de marcación IHQ para el gen supresor tumoral p53. Algunos autores ya han sugerido modificaciones para las próximas ediciones43–45.
. A) Se observan varias figuras de mitosis (flechas) en un campo de gran ampliación (HE, 400×). B) Confirmación inmunohistoquímica de las mitosis con el anticuerpo PHH3 (PHH3, 400×). C) El tumor muestra elevado índice proliferativo (8%, Ki-67, 200×). D) Inmunorreactividad nuclear extensa para p53 (p53, 200×).")
Características diagnósticas de los adenomas hipofisarios atípicos (ejemplo de un adenoma atípico de células nulas). A) Se observan varias figuras de mitosis (flechas) en un campo de gran ampliación (HE, 400×). B) Confirmación inmunohistoquímica de las mitosis con el anticuerpo PHH3 (PHH3, 400×). C) El tumor muestra elevado índice proliferativo (8%, Ki-67, 200×). D) Inmunorreactividad nuclear extensa para p53 (p53, 200×).
Se han propuesto otros marcadores de transformación de los AH, como la catepsina B o la metaloproteasa-9 (MMP-9), evaluación de la actividad proliferativa usando marcadores antiapoptóticos como el bcl-2, análisis de índices de DNA topoisomerasa II-alfa, expresión de ciclooxigenasa II, detección de la expresión de telomerasa o, estudios con galectina-3. Pero, desafortunadamente, ninguno ha mostrado mayor utilidad como marcador del comportamiento biológico, que el subtipo histológico basado en el contenido hormonal y la estructura celular que, continúan siendo los mejores marcadores predictivos independientes del comportamiento agresivo. La ausencia del gen p53, la disminución en la expresión de células folículo-estrelladas, del gen nm23, las anomalías de p27, p21, el análisis de vascular endotelial growth factor, CD34, fibroblast growth factor receptor 4, pituitary tumor transforming gene, deleciones en el cromosoma 11 y perfil de microRNAs, también se han propuesto para valorar la agresividad de estos tumores, pero no se han contemplado hasta ahora dentro de los criterios para la clasificación de los AH46–48. Es recomendable un seguimiento más estrecho en los pacientes que presenten este tipo de adenomas.
Carcinomas hipofisariosLos carcinomas hipofisarios son muy raros2,49, representando menos del 1% de todas las neoplasias hipofisarias50,51. Se definen por la presencia de metástasis cerebromedulares y/o sistémicas. No existen criterios morfológicos para distinguir un adenoma localmente agresivo de un carcinoma cuando el tumor está limitado a la silla turca. El desarrollo de carcinoma hipofisario de un adenoma es excepcional y en la actualidad se carece de datos sobre esa secuencia52. La mayoría de los carcinomas hipofisarios son macroadenomas invasivos hormonalmente activos, representando los prolactinomas y los tumores secretores de ACTH dos terceras partes de los mismos50. A diferencia de los AH, los carcinomas muestran siempre sobreexpresión IHQ para la proteína p5350,53. Tras la confirmación de malignidad, el pronóstico es reservado, con supervivencia de un año en dos tercios de los pacientes54.
Oncocitomas fusocelularesLas características clínicas y de neuroimagen de este tumor son inespecíficas, y el diagnóstico se basa en gran medida en sus características histopatológicas. Clínicamente son indistinguibles de los adenomas no funcionantes, y los pacientes pueden presentar signos y síntomas de hipopituitarismo y alteraciones visuales55.
Histológicamente, como su nombre indica, se caracterizan por presentar una apariencia celular fusiforme y oncocítica. A diferencia de los AH, carecen de inmunorreactividad para marcadores neuroendocrinos y hormonas hipofisarias. Las células tumorales son inmunorreactivas para el antígeno de membrana epitelial, vimentina, bcl-2, S-100 y galectina-3; no suelen expresar o solo expresan focalmente proteína ácida fibrilar glial55–58. La observación relativamente reciente del factor de transcripción de la tiroides (TTF-1) en estos tumores, así como en pituicitos normales y otros tumores de la región hipofisaria, entre los que se incluyen pituicitomas y tumores de células granulares de la neurohipófisis, plantea la posibilidad de que todos ellos puedan representar variantes diferentes derivadas de un origen patogénico común59,60, y que el origen de este tumor no sean las células folículo-estrelladas de la hipófisis anterior como originalmente se pensaba57.
Tumores de la neurohipófisisPituicitomasLos pituicitomas (previamente llamados astrocitomas de la hipófisis posterior o infundibulomas) son tumores raros; su comportamiento clínico preciso no ha sido bien caracterizado. En la mayoría de los casos parecen comportarse como tumores de bajo grado de malignidad, con cierta tendencia a recidivas después de una escisión subtotal61.
Morfológicamente, están compuestos por células piloides alargadas dispuestas en fascículos, en un patrón que se asemeja al astrocitoma pilocítico (sin embargo, a diferencia del astrocitoma pilocítico la mayoría de pituicitomas carecen del patrón bifásico, las características fibras de Rosenthal y los cuerpos granulares eosinófilos). En el estudio IHQ, los pituicitomas no muestran inmunorreactividad para marcadores neuroendocrinos ni hormonas hipofisarias. Las células tumorales son típicamente positivas para vimentina y proteína S-100; también son inmunorreactivas para bcl-2 y TTF-162. Aunque la mayoría de los pituicitomas expresan proteína ácida fibrilar glial, la marcación puede ser variable e incluso ausente56,57,59.
Tumores de células granularesLos tumores de células granulares son tumores raros (alrededor de 60 casos reportados en la literatura63), generalmente incidentales encontrados en autopsias de adultos. Habitualmente son neoplasias benignas de crecimiento lento, aunque han sido descritos algunos casos con un comportamiento clínico más agresivo63.
Histológicamente, están compuestos por células poligonales grandes con abundante citoplasma granular (fuertemente positivo para PAS con diastasa), núcleo redondo con cromatina delicada y nucléolo uniforme. A nivel IHQ, la mayoría de los tumores son inmunorreactivos para NSE y CD68 y, similar al pituicitoma y oncocitoma fusocelular, también son fuertemente positivos para TTF-160. A diferencia de los que surgen en el sistema nervioso periférico, apenas una minoría de los encontrados en la región selar son positivos para S-10063.
Otras lesiones y tumores de la región selarMetástasisLas metástasis a la glándula hipofisaria pueden verse en el 1% de los especímenes quirúrgicos, aunque la incidencia de metástasis encontrada en hipófisis de autopsia puede ser mayor8,64. Aunque se informó que los tumores que provocan metástasis en la hipófisis comprenden hasta el 28% en series de autopsias, la mayoría de los tumores metastásicos son clínicamente asintomáticos65. La hipófisis posterior está implicada con más frecuencia que la anterior (en ocasiones presentando signos y síntomas de diabetes insípida). Los cánceres de mama y pulmón son las neoplasias primarias más comunes que provocan metástasis en la hipófisis65.
CraneofaringiomasRepresentan aproximadamente el 3% de todas las neoplasias intracraneales y cerca del 10% de los tumores de la región selar2,66. La mayoría surgen en la infancia y adolescencia (entre 5 y 15 años). En estudios de neuroimagen, los craneofaringiomas son lesiones típicamente calcificadas, sólidas, quísticas (o sólido-quísticas) con una compleja apariencia lobular.
Histológicamente, los craneofaringiomas muestran un complejo y característico patrón de crecimiento epitelial, siendo clasificados como tumores de grado I según los criterios de la OMS66. La clasificación de la OMS identifica dos variantes: adamantinomatosa (caracterizada por epitelio estratificado con una disposición en empalizada de las células basales, formación de queratina «húmeda», cambios microquísticos y una expresión nuclear aberrante de beta-catenina en hasta el 95% de los casos) y papilar (caracterizada por epitelio estratificado pavimentoso simple no queratinizante que reviste un estroma de tejido conjuntivo, usualmente formando estructuras seudopapilares)66, aunque a veces, estos tumores pueden expresar ambos patrones de crecimiento en proporciones variables.
Lesiones inflamatoriasLas enfermedades inflamatorias primarias de la glándula hipofisaria son poco comunes y pueden imitar tumores selares. La hipofisitis ha sido clasificada en tres categorías basadas en la presentación clínico-patológica: 1) hipofisitis linfocítica autoinmune (la más común y clínicamente relevante), 2) hipofisitis granulomatosa y 3) hipofisitis xantomatosa67,68. Los linfocitos no están normalmente presentes en la adenohipófisis, por lo que un número significativo de células inflamatorias en la glándula hipofisaria es siempre una condición patológica. La hipofisitis linfocítica es una entidad rara que afecta con más frecuencia a mujeres en la última fase del embarazo o en el período posparto69, siendo muy raro en hombres67,68,70. Se cree que tiene una base autoinmune, una vez que se han demostrado anticuerpos contra las células hipofisarias y asociación de otras enfermedades endocrinas o inmunes en cerca del 20% de los pacientes69. Microscópicamente se caracteriza por una infiltración linfoplasmocítica de la adenohipófisis, que en etapas posteriores de la enfermedad puede provocar una atrofia del parénquima glandular, grado variable de fibrosis y agregados linfoides residuales.
En raras ocasiones, la glándula hipofisaria también puede estar afectada secundariamente por procesos inflamatorios o infecciosos sistémicos (como sarcoidosis o tuberculosis), por lo que deben ser excluidos antes de un diagnóstico final de hipofisitis primaria.
Otras lesiones del área selarUna gran variedad de otras lesiones y tumores pueden afectar la hipófisis y región selar. Estos incluyen tumores originados en la duramadre y revestimiento de la silla turca (como el meningioma), las estructuras óseas (como el cordoma) y la médula ósea (como el plasmocitoma o la histiocitosis de células de Langerhans), por lo que debemos tener siempre presente que no todo es adenoma.
ConclusionesEl patólogo tiene un papel clave en el equipo multidisciplinar que trata a los pacientes con patología hipofisaria y, para lograr un diagnóstico preciso, le resulta fundamental una estrecha colaboración con el restante equipo médico.
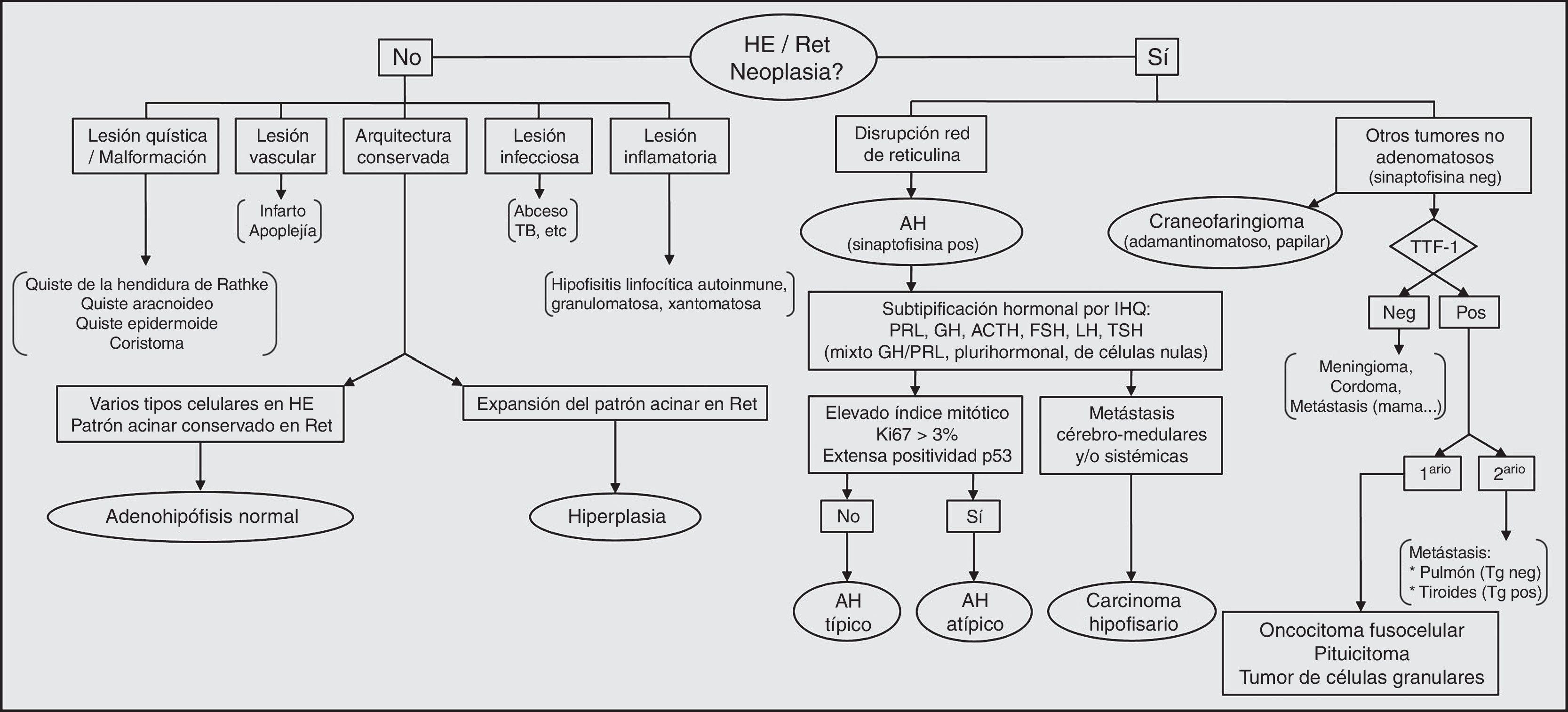
Se describe un diagrama de flujo para la patología hipofisaria más frecuente (fig. 4), que incluye algunos marcadores IHQ de diferenciación celular así como de pronóstico. Con nuevos biomarcadores para AH agresivos y nuevos datos sobre anomalías genéticas asociadas con la patogénesis tumoral hipofisaria, se prevé que sea aclarada la relación entre adenomas típicos, atípicos y carcinomas, lo que permitirá mayor precisión diagnóstica y pronóstica.
![Algoritmo diagnóstico en la patología hipofisaria. AH: adenoma hipofisario; HE: hematoxilina-eosina; IHQ: inmunohistoquímica; neg: negativo; pos: positivo; Ret: reticulina; TB: tuberculosis; Tg: tiroglobulina; TTF-1: factor de transcripción de la tiroides; Adaptada de: Ortiz S, Tortosa F. The mind map in pituitary/sellar pathology - A practical approach. [Comunicación]. XXXI International Congress of the International Academy of Pathology. 25-29 de Septiembre de 2016. Colonia, Alemania.](https://static.elsevier.es/multimedia/25300164/0000006400000003/v1_201703170143/S2530016416300088/v1_201703170143/es/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNf23BlP/DWEpPOpuWlfofnyqxn4xVHcGY2cyFujnbUAmLxF48+d8JnEaosD0RrpPP8znX6W9O+9RPKUxiwmOD77LcoyMTmWrbxhWepkRGFVehWSphuwok7jhjib9pJIsgbH5upQvgJBsRnDRwt6pbazsElKAzZ3EktVH6ffZqjXL1WWrmRGq1U4tgPqR2yK7ArHNtsGhjLe/r8wM9A6kxl5GohLsvv7cdLi7g05YhebQhiD2TnyMXQZqGvzPO83QAj3PT1xUOzn68lhooOzQFyD "Algoritmo diagnóstico en la patología hipofisaria. AH: adenoma hipofisario; HE: hematoxilina-eosina; IHQ: inmunohistoquímica; neg: negativo; pos: positivo; Ret: reticulina; TB: tuberculosis; Tg: tiroglobulina; TTF-1: factor de transcripción de la tiroides; Adaptada de: Ortiz S, Tortosa F. The mind map in pituitary/sellar pathology - A practical approach. [Comunicación]. XXXI International Congress of the International Academy of Pathology. 25-29 de Septiembre de 2016. Colonia, Alemania.")
Algoritmo diagnóstico en la patología hipofisaria. AH: adenoma hipofisario; HE: hematoxilina-eosina; IHQ: inmunohistoquímica; neg: negativo; pos: positivo; Ret: reticulina; TB: tuberculosis; Tg: tiroglobulina; TTF-1: factor de transcripción de la tiroides; Adaptada de: Ortiz S, Tortosa F. The mind map in pituitary/sellar pathology - A practical approach. [Comunicación]. XXXI International Congress of the International Academy of Pathology. 25-29 de Septiembre de 2016. Colonia, Alemania.
*La hipófisis puede verse afectada por una amplia gama de lesiones, que a veces cursan con características clínico-radiológicas similares. Se debe tener siempre presente que puede no tratarse de un adenoma.
*Para una evaluación patológica adecuada de los AH se requiere un extenso estudio IHQ y, en algunos casos, microscopía electrónica.
*Algunos AH son intrínsecamente agresivos; el subtipo histológico basado en el contenido hormonal y la estructura celular continúan siendo los mejores marcadores predictivos del comportamiento agresivo.
*El patólogo tiene un papel clave en el equipo multidisciplinar de pacientes con tumores de la región selar.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
