





Recently, there have been advances in understanding of the changes that occur in the hypothalamic-pituitary-adrenal axis during the different stages of critical disease. Such advances have led to a paradigm change, so that the aforementioned adaptations are no longer considered the result of adrenal axis activation, but a consequence of decreased cortisol metabolism illness. Knowledge of this new pathophysiological bases should lead to reconsider the diagnosis and treatment of adrenal insufficiency in critically ill patients, a condition poorly understood to date.
En los últimos años se han producido avances en el conocimiento de los cambios que experimenta el eje suprarrenal durante las distintas fases de la enfermedad crítica. Dichos avances han cristalizado en un cambio de paradigma, de modo que las referidas adaptaciones ya no se consideran resultado de la activación del eje a nivel hipotalámico, como se ha considerado tradicionalmente, sino fruto de una disminución en el metabolismo periférico del cortisol. Estos nuevos datos obligan a reconsiderar el diagnóstico y tratamiento de la insuficiencia suprarrenal del enfermo crítico, una entidad hasta ahora escasamente comprendida.
During critical disease, the most severe form of physical stress, significant metabolic and endocrinological changes occur, initially intended to promote adaptation of the body to its fight for life. Advances in intensive medicine have substantially improved prognosis of previously fatal conditions. This allows many patient to survive for long time periods at intensive care units (ICU) and to reach the so-called chronic stage of critical diseases.
In recent years, new evidence has been obtained on the pathophysiological changes that occur during body response to stress, which may have significant therapeutic implications Thus, it is now known that two markedly different phases exist during critical disease: the first phase, or acute phase, comprise the first few days of the disease, while the second phase, or chronic phase, would only be reached by patients who do not recover (or die) in the first few days of the disease. In the first phase, metabolic changes occur mainly in peripheral tissue. These1,2 changes are aimed at promoting body adaptation to the disease. In the second phase, changes of a central origin occur in the hypothalamic-pituitary axes. These changes, unlike those occurring in the acute phase, may contribute to aggravate the catabolic condition of patients and hinder recovery.1,3 If this was true, hormone treatments could help improve disease prognosis.4
Although these considerations are valid for all hypothalamic-pituitary axes, in recent years, the most marked advances have been made in understanding of adrenal axis changes.
Relevance of the adrenal axis during critical diseaseExposure of the body to a stressor of adequate intensity triggers the massive released of different mediators and hormones with different time sequences. The first wave of hormonal response occurs in a few seconds and includes catecholamine release by the nervous system as the most significant and earliest event.5 A second wave, occurring somewhat later (in minutes), mainly results in increased plasma glucocorticoid levels. Catecholamines released in the first wave induce cardiovascular activation (increases in heart rate, cardiac output, and blood pressure), which is considered indispensable for surviving critical disease. They also promote blood flow diversion to the tissues most actively fighting the stressor, and contribute to nutrient mobilization from their deposits to make them available to such tissues.
Glucocorticoids have a more complex role in stress situations, because they regulate a wide variety of physiological functions. It has long been known that cortisol requirements are increased in critical conditions, and6–8 that the impossibility to increase cortisol availability in such situations, as occurs in patients with structural disease of the hypothalamic-pituitary-adrenal axis (HPA) or on long-term corticosteroid therapy, increases risk of mortality.6
Overall, glucocorticoid actions are intended to promote the fight against the stressor, or to counteract an excessive defense reaction to the stressor that could be detrimental to the body. The former include permissive actions (occurring early, when cortisol levels have not increased yet), and stimulating actions (occurring at a later time in the presence of cortisol levels increased in response to stress). Thus, for example, hemodynamic effects of glucocorticoids during stress result from their permissive action on catecholamines, while their metabolic effect result from both permissive and stimulating actions. Actions of glucocorticoids aimed at protecting the body from an excessive defense reaction are called suppressing actions and particularly include those exerted on the immune system. In this case, the initial effect of glucocorticoids is permissive, to promote immune and inflammatory response. Subsequently, when plasma glucocorticoid levels are increased, the effect becomes clearly suppressive, in order to protect the body from excess immune activation.5,9
It is therefore understandable that availability of adequate cortisol levels is of utmost importance to overcome stress situations.
Cortisol insufficiency during critical disease may result in structural damage in any point of the HPA axis occurring either before critical disease or during the disease as a consequence of its complications (e.g. adrenal hemorrhage or thrombosis resulting from coagulation disorders). In addition, some treatments commonly used at the ICUs, such as ketoconazole or etomidate, may lead to inadequate cortisol10 production because of their inhibitory effect on steroid synthesis. In both cases, cortisol production by the adrenal gland may not be adequate to cover patient requirements, even in the absence of physical stress. This would therefore be a total11,12adrenal failure, which is outside the scope of this review.
On the other hand, the critical disease itself may induce a functional, transient disorder of the adrenal axis that causes inadequate cortisol production. This disorder, traditionally called relative adrenal failure, is now known as adrenal failure of critical patients.13–15 This is a controversial condition that has been increasingly understood in recent years, and which is the focus of this review.
Response of the HPA axis to stress in critical patients: new conceptsIt has traditionally been considered that, as regards the adrenal axis, body response to aggression is characterized by central activation of the HPA axis. However, as noted above, two phases with markedly different causes and consequences are now distinguished.
Acute phaseThe acute phase starts when aggression to the body occurs, lasts a few days, and is characterized by increased plasma cortisol levels. This increase in cortisol levels during the first days of critical disease was traditionally considered the result of increased cortisol production due to central activation of the HPA, which would lead to increased secretion of CRH and, secondarily, ACTH.
This conception of response of the HPA axis during critical disease has been questioned based on the evidence accumulated in recent years. In 1995, Vermes et al. demonstrated that, contrary to what was expected, ACTH levels were only transiently elevated in critical patients.16 More recently, Boonen et al. showed that critical patients had ACTH levels lower than healthy subjects from the first day of ICU admission, despite which they maintained higher cortisol levels.17 This dissociation between cortisol and ACTH levels was not consistent with the classical concept of central activation of the HPA axis and suggested involvement of factors other than ACTH in regulation of cortisol production during critical disease. Among these factors, various cytokines, as well as catecholamines and neuropeptides, were postulated.18,19 However, quantification by Boonen et al. of cortisol production and metabolism using isotopic techniques revealed an unexpected finding: cortisol production was only slightly increased as compared to healthy individuals. Moreover, such increase was only found in patients with more marked inflammation, while in the great majority of critical patients cortisol production, against all odds, was not different from that of healthy individuals. By contrast, cortisol metabolism or clearance was markedly decreased in all patient groups.17 In agreement with these findings, Boonen et al. showed profound inhibition of the expression and activity of the liver enzymes 5α-reductase and 5β-reductase and the renal enzyme β-hydroxysteroid dehydrogenase,17 all of them responsible for conversion of cortisol to inactive metabolites. Potential mediators of this inhibition proposed include bile acids, whose plasma levels are markedly increased during critical disease and positively correlates to cortisol levels.17 In addition, bile acids are known inhibitors of transcription and activity of hepatic reductases,20–22 and their levels are inversely correlated to expression, tissue levels, and activity of these enzymes.17
Subsequent research by the same authors showed that the decrease in ACTH levels found in this phase of disease resulted from negative feedback exerted by hypercortisolism, which would induce a decreased amplitude of ACTH secretory pulses by pituitary corticotroph cells.23
Teleologically, reduction of cortisol metabolism may be interpreted as a more economical way (as compared to increased production) for the body to increase cortisol availability in critical situations. On the other hand, this mechanism results in proportionally greater increases in cortisol levels in organs responsible for its inactivation than in the bloodstream. This would in turn promote greater availability of cortisol in key organs such as liver and kidney, while minimizing availability in other tissues such as the immune and musculoskeletal systems or the brain, in which excess exposure to cortisol could have adverse effects. In this regard, selective regulation of glucocorticoid receptor expression in the different tissues has also been postulated. This would allow for focusing cortisol actions on the organs of interest and for minimizing its adverse effects. Thus, for example, the glucocorticoid receptor of granulocytes has been found to be decreased in children with sepsis, which would allow development of an innate immune response despite hypercortisolism.24 However, regulation of cortisol receptor in critical diseases is poorly known and requires further research.
Chronic phaseIf therapeutic and support measures during the acute phase of critical disease are successful, patients will improve and overcome the critical situation in a few days. By contrast, if patients survive the acute phase of disease but do not recover from the disease, they will enter the so-called chronic phase of critical disease. As demonstrated by Boonen et al., this phase is characterized by progressive adrenal gland atrophy. These authors performed postmortem studies of critical and non-critical patients and found that adrenal glands of patients who died after long stays at ICUs had more disorganized architectures, lower weights, and lower cholesterol ester levels as compared to patients dying shortly after admission and those who died at other units providing less intensive care.25 Although the study design did not allow for measuring plasma ACTH levels, the finding of decreased expression of genes regulated by this hormone and the parallelism between histological findings and those made in mice with POMC mutation (and, thus, ACTH-deficient)26 suggested that persistently low ACTH levels in critical patients were involved in the genesis of adrenal atrophy.
On the other hand, Barquist et al. showed an inverse correlation between plasma cortisol levels and time of ICU stay.27
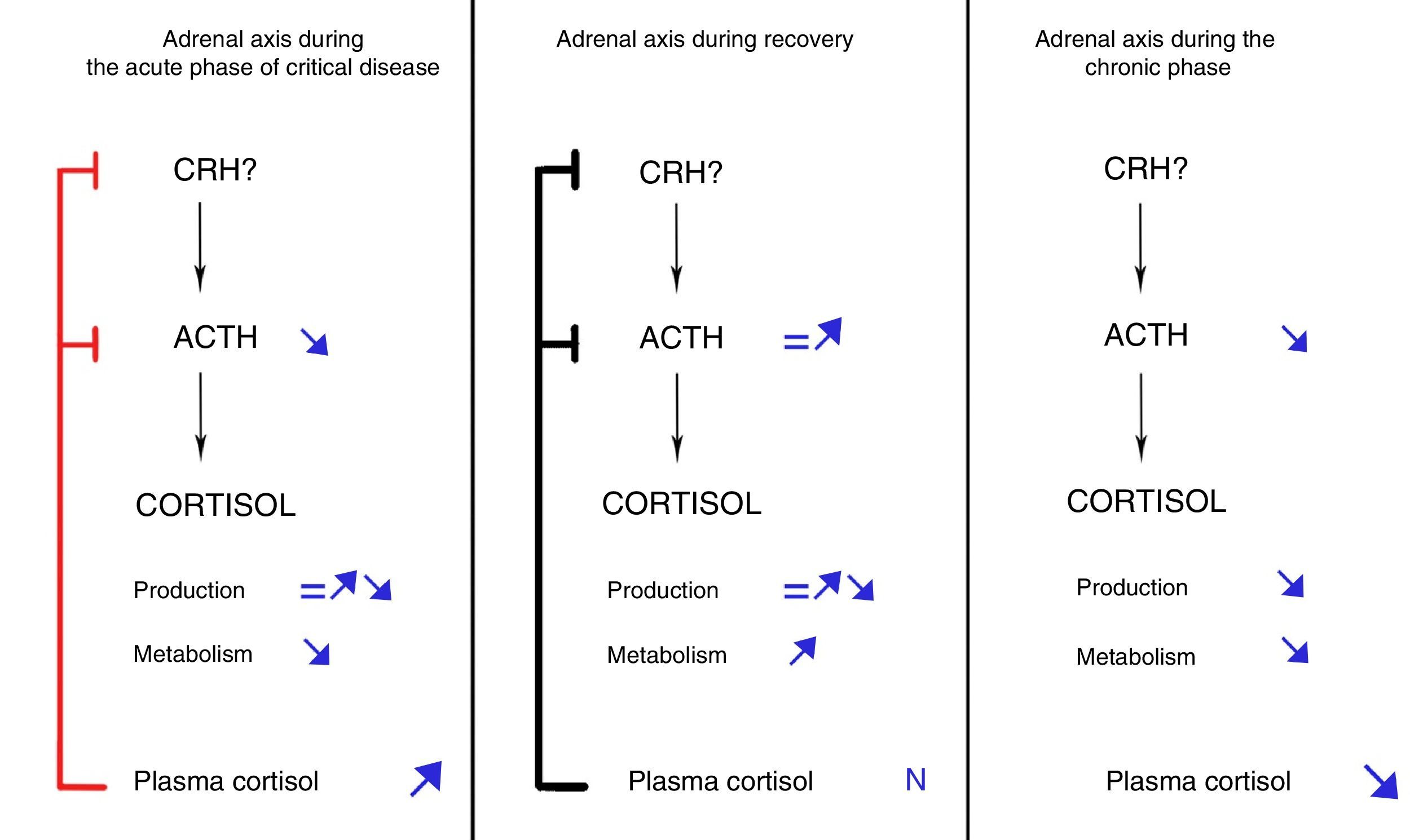
These findings led to propose the sequence of facts shown in Fig. 1: if a critical patient does not recover in the first few days of disease, inhibition of ACTH secretion started in the acute phase would prolong in time due to maintenance of the critical condition. As the result, persistently low ACTH levels would induce adrenal gland atrophy, which would in turn lead to a gradual decrease in cortisol secretion.25 The net balance between cortisol increased derived from inhibition of its metabolism (that would persist in the chronic phase) and the decrease resulting from decreased production would gradually tilt in favor of the latter, resulting in a progressive decrease in plasma cortisol levels. This sequence of facts would explain the increase in the rate of symptomatic adrenal insufficiency as ICU stay becomes longer.27

Dynamic evolution of the pituitary-adrenal axis during critical disease. Modified from Boonen and van Den Berghe.28
By contrast, if patients overcome the critical situation in a few days after ICU admission, the processes responsible for inhibition of cortisol metabolism would revert, plasma cortisol levels would return to normal, and ACTH secretion would secondarily normalize, thus fully restoring adrenal function.
Does adrenal insufficiency actually occur in critical patients?It appears evident that increased cortisol availability in the acute phase of critical disease has an adaptive nature. By contrast, it does not appear to be so clear whether the decrease in plasma cortisol levels occurring in the chronic phase is also adaptive in nature (in which case, existence of adrenal insufficiency in critical patients would be questioned), is a harmful event that may hinder patient recovery, or is simply a marker of severity. This question may only be answered by intervention studies. In a study conducted on genetically manipulated animals to achieve inactivation of the scavenger receptor BI in adrenal cells (responsible for internalization of HDL cholesterol molecules to produce cortisol when cortisol requirements increase) and thus induce adrenal gland hyporesponsiveness to stress, Ai et al. showed greater mortality from sepsis in these animals as compared to normal animals, and that treatment with glucocorticoids was able to revert the increase in mortality.29 However, this experimental model is more representative of absolute adrenal insufficiency (defined as that resulting from damage to the HPA axis prior to critical disease) than of adrenal insufficiency of critical patients. On the other hand, Guzman et al. reported a series of 13 septic patients in whom mean cortisol level changed from 41μg/dL at ICU admission to 10μg/dL when retested one week later due to impossibility to discontinue treatment with vasoactive drugs. Administration of glucocorticoids to these patients allowed for discontinuation of vasopressor treatment and for ICU discharge after 1.5 days on average.30 The lack of a control group is an obvious limit to draw robust conclusions, but this study suggests a benefit derived from glucocorticoid therapy and the maladaptive nature of adrenal function changes in advanced stages of critical disease. However, this should be confirmed in the future in duly designed studies.
Diagnostic implicationsHemodynamic instability despite adequate fluid replacement and persistence of signs of inflammation with no evident cause are clinical data that should lead to suspect adrenal insufficiency in critical patients. Other data that may be helpful for diagnosis under normal conditions, such as electrolyte disorders, are of little value in critically ill patients. On the other hand, laboratory confirmation of adrenal insufficiency in critical patients is quite difficult and has traditionally been controversial. However, adequate identification of this disorder is important to be able to focus therapeutic intervention in those who suffer from it and to avoid overtreatment of those with no insufficiency. This is suggested by recent studies showing in animal models that, in the absence of adrenal insufficiency, glucocorticoid treatment is not only not beneficial, but may increase the risk of catabolic complications and infection, amongst others.29 Tests that assess the overall HPA axis, such as insulin-induced hypoglycemia, are not adequate and have not been validated in critical patients. Because of this, the variables traditionally used for diagnosis have been basal serum cortisol levels and maximum increase in serum cortisol after stimulation with 250μg of ACTH. In both cases, identification of abnormal values for critical patients has been based on their association to risk of mortality. As the mortality risk of a critical patient depends on multiple factors, including the degree of stress, and since basal plasma cortisol is correlated to the degree of stress, both high cortisol levels (reflecting a higher degree of stress) and low cortisol levels (which would reflect an inadequate cortisol response) have been associated to increased mortality.31 As the result, values proposed as normal for basal plasma cortisol levels in critical situations have been very disparate. Response to the ACTH test, apparently less dependent on the degree of stress, would theoretically be of greater value, although the results have also been poorly reproducible.32 Some authors have proposed use of the dose of ACTH 1μg to increase sensitivity of the test, but this has not been adequately tested yet.33 As of today, a great part of the lack of reproducibility of the above mentioned tests (and the resultant disparity in diagnostic criteria) could be explained by their use at different stages of the critical disease, in which pathophysiological changes in the HPA are very disparate in nature. According to the new concept of stress response, ACTH levels are decreased since the start of disease, but only critical patients with long duration of disease will experience decreases in plasma cortisol levels (greater the longer the duration of disease). Several diagnostic considerations may be made based on these facts: (1) low ACTH levels should not be considered indicative or suggestive of adrenal insufficiency (secondary in this case); (2) the possibility of adrenal insufficiency induced by the critical disease should only be envisaged in advanced disease stages; (3) the finding of low cortisol levels in the acute phase of critical disease should be interpreted to reflect organ damage in some HPA axis either prior to critical disease and not recognized, or resulting from disease complications (it would thus be the so-called absolute adrenal insufficiency). The normal range of cortisol levels in this first phase has not been defined yet. According to the Boonen et al. study,17 mean cortisol levels during the first week of ICU stay in patients with (state condition) was 16.8μg/dL. Both basal plasma cortisol levels and the ACTH stimulation test may be valid to diagnose adrenal insufficiency in critical patients as currently perceived, because adrenal gland atrophy in advanced disease stages is already evident. In both cases, the separation threshold between physiological and pathological levels should be established based on the benefits provided by glucocorticoid therapy and be considered as a diagnostic criterion. It should be noted, however, that the decrease in the cortisol-binding protein associated to critical disease34–36 leads to decreased total cortisol levels without affecting the free fraction (the only biologically active fraction), which complicates interpretation of total cortisol levels in critical conditions and may lead to false positive diagnosis. On the other hand, tests to measure the free fraction, which would theoretically be helpful to overcome this disadvantage, are unreliable,37–39 and are not currently available at most hospitals.
Therapeutic implicationsTo date, studies intended to show the benefit of steroid therapy in critically ill patients have provided conflicting, not conclusive results.40–42 The lack of reproducibility in these studies is likely to be due again to a design based on the concepts prevailing in the past on changes in the adrenal axis in response to stress, which did not contemplate the dynamic nature of such changes. This fact may explain, for example, that studies assessing response to treatment in the early stages of disease (when adrenal insufficiency is highly unlikely) found no benefit, while studies in which treatment was administered in late stages did find beneficial effects.30
The lack of well-established criteria for diagnosis of adrenal insufficiency in critical patients makes selection of patients candidate to treatment difficult. Until such diagnostic criteria are established in further studies, Boonen et al., based on the dynamic response of the adrenal axis in critical situations, have proposed as candidates to steroid treatment those critical patients who meet the following criteria: (1) compatible symptoms (vasoactive drug dependence mainly); (2) ICU stay longer than 6 days; (3) basal plasma cortisol levels decreasing to values less than 6μg/dL; and (4) increased cortisol levels after stimulation with less than 6μg/dL of ACTH.28 The hydrocortisone dose recommended and traditionally used to treat adrenal insufficiency in critical patients has ranged from 200 to 300mg/day. Such recommendation was based on the belief that the increase in plasma cortisol levels seen in critical disease resulted from a marked increase in cortisol production. In the light of new knowledge, this dose probably results in excessively high cortisol levels, because in critical situations reductions occur not only in endogenous cortisol metabolism, but also in metabolism of drug preparations, which causes a substantial prolongation of their half-life.17 These high doses may be associated to harmful effects on some organ systems that may limit treatment benefits. Based on isotopic studies to investigate daily cortisol production in critical situations, some authors suggested that daily doses of 60mg of hydrocortisone would be adequate in such situations.28 Dosage should be tapered as soon as the patient starts to recover.
These considerations about steroid dosage also apply to critical patients who receive during their stay at the ICU steroid treatment for indications other than adrenal insufficiency.
The main diagnostic and therapeutic implications are summarized in Table 1.
Main diagnostic and therapeutic implications.
| Adrenal insufficiency should only be suspected in critically ill patients with long ICU stays (longer than one week) |
| Low ACTH levels should not be considered suggestive of this condition |
| In more severe cases, use of basal serum cortisol levels may be sufficient for diagnosis. In doubtful cases, ACTH stimulation test may be used |
| It is not known which levels of basal or stimulated cortisol are indicative of adrenal insufficiency in critical patients. Basal serum cortisol levels less than 6μg/dL have been suggested |
| The most adequate hydrocortisone dose for replacement therapy should probably be lower than the dose currently used A dose of 60mg daily (instead of 300mg daily) has been proposed |
Future studies designed taking into account the new knowledge about the pathophysiology of the adrenal axis in critical diseases should serve to establish the indications for treatment, as well as the optimum doses to be used in these conditions.
ConclusionsResponse to adrenal axis disease is dynamic and consists of two phases with different causes and consequences. Changes in the acute phase are adaptive and require no treatment, while changes occurring in the chronic phase may contribute to worsen patients prognosis and would warrant steroid treatment. There is currently no solid evidence to support diagnostic and therapeutic recommendations, because there are still many aspects to be elucidated. Among such aspects, the impact of declining cortisol levels on critical disease (or patient) prognosis and the factors contributing to or enhance this decrease should be confirmed. The threshold levels of plasma cortisol (or other biochemical parameters) that allow for identification of patients amenable to treatment should also be established. Finally, the optimum replacement doses that allow for minimizing the risks associated to overtreatment should be determined. These questions should be addressed in studies designed taking into account the new understanding of response of the adrenal axis in critical situations.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Mateos Moreno L, Palacios García N, Estrada García FJ. Insuficiencia suprarrenal en el enfermo crítico: nuevos conceptos etiopatogénicos e implicaciones terapéuticas. Endocrinol Diabetes Nutr. 2017;64:557–563.
