






La enfermedad de von Hippel-Lindau (VHL) es un trastorno de herencia autosómica dominante de alta penetrancia que presenta afectación de múltiples órganos, con expresión variable: sistema nervioso central, retina, riñones, páncreas, etc. Para su diagnóstico se utilizan los criterios de Melmon y Rosen así como el estudio de las alteraciones genéticas.
La afectación pancreática consiste en tumores quísticos (TQP), tumores neuroendocrinos (TNE) y adenocarcinomas fundamentalmente. El manejo de las lesiones quísticas difusas suele ser conservador, pero su asociación con TNE puede condicionar la técnica quirúrgica en caso de intervención.
Presentamos el caso de una paciente con afectación difusa del páncreas por TQP asociado a un TNE que aumentó de tamaño durante el seguimiento.
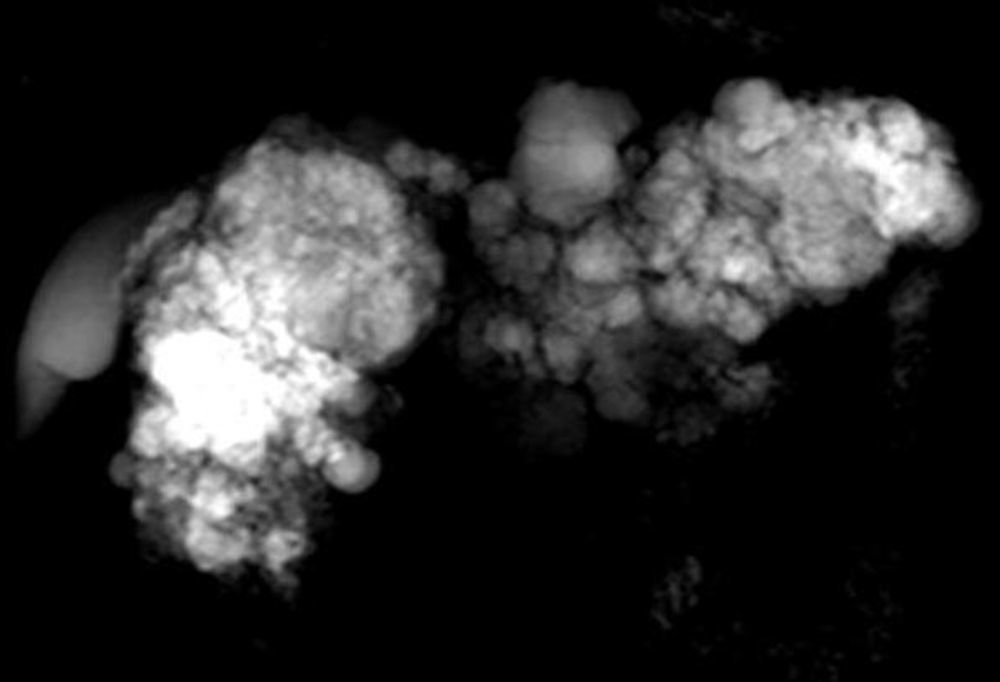
Observación clínicaSe trata de una mujer de 36 años, con antecedentes de diabetes mellitus insulinodependiente tipo 1, diagnosticada recientemente de enfermedad de VHL a raíz del hallazgo casual, en una resonancia magnética (RM) de columna vertebral, de una serie de lesiones intraabdominales que indicaban el diagnóstico. El estudio genético confirmó el diagnóstico encuadrando a la paciente dentro del tipo 1. Las lesiones descubiertas correspondían a un quiste renal de 43×30mm en polo superior izquierdo, 3 hemangiomas hepáticos de 1-2cm y la existencia de un páncreas completamente sustituido por múltiples lesiones sólido-quísticas (fig. 1) de diferentes tamaños, algunas con contenido hemorrágico y una de ellas sólida localizada en cuello pancreático, de 35mm de diámetro máximo, compatible con un tumor neuroendocrino, sin afectación del conducto de Wirsung ni de la vía biliar. Así mismo se descubrió la existencia de 4 lesiones cerebelosas, de entre 6 y 23mm, compatibles con hemangioblastomas. El fondo de ojo era normal.

A los 6 meses del diagnóstico, la paciente presentó sintomatología de hipertensión intracraneal, por lo que fue derivada a otro centro e intervenida de las lesiones cerebelosas con éxito y sin complicaciones.
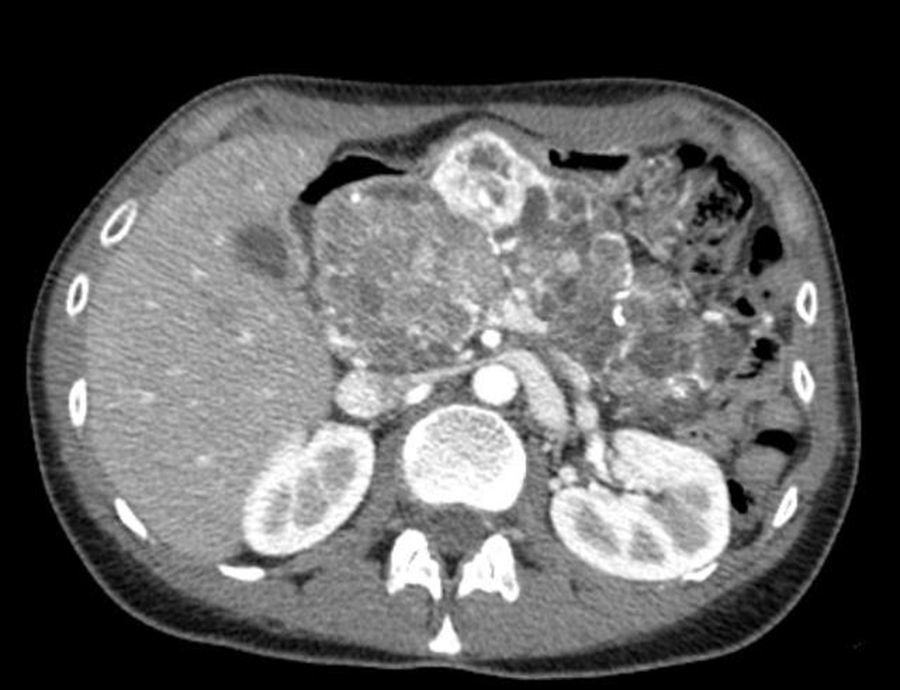
Tres meses después de esta intervención nos es remitida nuevamente para seguimiento. La paciente llevaba buen control de su diabetes (HbA1c 7%), pero unas semanas antes había consultado por ciertas molestias abdominales y deposiciones pastosas. La elastasa pancreática fecal tenía valores inferiores a 15μg/g de heces (VN > 20μg/g de heces). Con la administración de un preparado de enzimas pancreáticas vía oral los síntomas desaparecieron. En una TC de seguimiento realizada a los 10 meses del diagnóstico se aprecian las mismas lesiones pancreáticas, pero el nódulo sólido con realce periférico nodular situado en el cuello y correspondiente a un TNE presenta aumento del tamaño, con un diámetro máximo de 46mm (fig. 2).

Se realizó un estudio hormonal completo siendo normales los valores de cromogranina A, polipéptido pancreático, VIP, glucagón, enolasa, gastrina y catecolaminas, siendo el 5OH-indolacético el único que presentaba un discreto aumento (12,9mg/24h; VN hasta 10mg/24h). Tanto el CEA como el CA 19.9 eran normales también.
Debido al crecimiento y las características radiológicas de la lesión se decidió la intervención quirúrgica. Los hallazgos intraoperatorios coincidían con los radiológicos encontrando una glándula pancreática voluminosa, completamente ocupada por lesiones quísticas y un nódulo sólido en cuello. Se realizó una duodenopancreatectomía total (fig. 3) y reconstrucción en una sola asa. El postoperatorio fue satisfactorio y le fue dada el alta al octavo día.

El estudio anatomopatológico confirmó la existencia de un TNE pancreático de 2,5cm de diámetro sin actividad mitótica excepto en un pequeño foco de 2mm con KI-67 del 60%; además existían cordones neuroendocrinos intersticiales sin actividad proliferativa. El resto del parénquima estaba ocupado por una poliquistosis. Ninguno de los ganglios aislados presentaba infiltración neoplásica.
Ocho meses después en una TC de control se descubrió la aparición de múltiples metástasis bilaterales hepáticas de origen neuroendocrino que causaron el fallecimiento de la paciente un año después.
DiscusiónLa enfermedad de VHL es una enfermedad hereditaria autosómica dominante con alta penetrancia y expresión variable. Se caracteriza por neoplasias que afectan a múltiples órganos. Las manifestaciones más comunes son hemangioblastomas de SNC, angiomas retinianos, carcinomas de células renales, feocromocitoma, lesiones pancreáticas y quistes del epidídimo1.
La enfermedad aparece en 1:36000 recién nacidos vivos y presenta una alta penetrancia dependiente de la edad, llegando al 90% a los 60 años. Se debe a una mutación en el gen de supresión tumoral VHL que se localiza en el cromosoma 3p252. Este gen contiene 3 exones, que como ocurre en la mayoría de síndromes hereditarios, expresará diferentes genotipos-fenotipos en función del tipo de mutación que se produzca.
Existen 3 subtipos clínicos y moleculares de la enfermedad de VHL2–4 que se muestran en la tabla 1.
Nuestra paciente se incluyó dentro del subtipo 1, por lo que el riesgo de sufrir feocromocitoma era bajo, y se comprobó con niveles de metilnefrinas normales.
La principal causa de muerte en pacientes con VHL son las complicaciones relacionadas con la aparición de carcinoma de células renales y hemangioblastoma del SNC2. Aun así, debido a la alta prevalencia de desarrollar distintos tipos de tumores en otros órganos, el cribado y seguimiento de otros territorios en estos pacientes debe ser muy estrecho.
El diagnóstico de la enfermedad se establece mediante los criterios diagnósticos descritos por Melmon y Rosen en 19645,6, que se definen como la presencia de un hemangioblastoma en SNC y otra lesión de VHL, o bien una lesión de VHL en un familiar de un paciente afectado de VHL.
En la mayoría de los casos, el diagnóstico se realiza por la aparición de hemangioblastomas retinianos, que normalmente es el primer signo de diagnóstico de VHL. Son lesiones esporádicas en la población general que, en pacientes afectados por VHL, se manifiestan a edades más tempranas, multifocales y bilaterales. En nuestra paciente, el diagnóstico no se hizo de esta forma, ya que se inició el estudio a raíz del hallazgo de lesiones quísticas abdominales en una prueba de imagen.
El hemangioblastoma cerebeloso es la lesión más frecuente y suele aparecer de manera precoz en pacientes con VHL. Aunque se trata de lesiones benignas, su clínica se relaciona con su tamaño y el efecto de compresión que producen. Tienen indicación quirúrgica cuando aparece sintomatología y a diferencia de nuestra paciente, que quedó asintomática tras la exéresis del hemangioblastoma cerebeloso, el déficit neurológico normalmente no es reversible después de la cirugía y el resultado final depende del estado preoperatorio4.
El riesgo de carcinoma renal está muy aumentado respecto a la población general (28-45%), presentándose a edades más tempranas (30-36 años)7. Las lesiones renales más frecuentes son los quistes renales, que se deben considerar una lesión premaligna, y el carcinoma renal de células claras, normalmente multicéntricos y bilaterales. El diagnóstico y seguimiento se debe realizar mediante TC o RM cada 6 meses que, en pacientes pertenecientes a familias diagnosticadas de VHL, deberá comenzar a los 15 años y se ha relacionado con una disminución en la edad del diagnóstico8. El tratamiento quirúrgico se indica a partir de lesiones mayores de 3cm.
La evolución de la enfermedad renal en nuestra paciente no fue agresiva, ya que a pesar de presentar lesiones quísticas renales, estas no se malignizaron ni marcaron el pronóstico de la enfermedad, como es característico en estos pacientes.
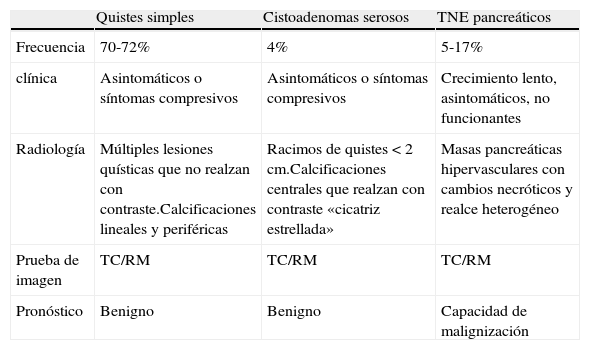
La afectación pancreática en pacientes con VHL es común (50-70%)3 siendo la mayoría lesiones quísticas (70%) y cistoadenomas (4%). Estas lesiones no tienen potencial maligno, pero pueden reemplazar la glándula pancreática hasta causar insuficiencia endocrina y exocrina como ocurrió en nuestra paciente, que se manifestó con DM por ausencia de glándula pancreática sustituida por lesiones quísticas. En otras ocasiones, estas lesiones pueden causar síntomas secundarios a la compresión del intestino o conducto biliar7,9. El diagnóstico diferencial mediante pruebas de imagen es esencial siendo la TC y la RM las técnicas de elección (tabla 2). Los quistes simples se caracterizan por presentar múltiples lesiones quísticas que no realzan con contraste con paredes finas en «panel de abeja». Si existen calcificaciones, son lineales y situadas en la periferia del quiste. Por otro lado, los cistoadenomas se presentan como racimos de quistes menores de 2cm con calcificaciones centrales que realzan contraste, lo que se describe como «cicatriz estrellada». Se ha descrito una variante específica para la enfermedad de VHL que se caracteriza por la aparición de cistoadenomas serosos que afectan a toda la glándula10. Neumann et al.11 recomiendan una actitud terapéutica conservadora en este tipo de lesiones manteniendo el seguimiento a largo plazo cada 6 meses e indicar la cirugía si aparecen complicaciones.
Diagnóstico diferencial de las lesiones quísticas del páncreas en Von Hippel-Lindau
| Quistes simples | Cistoadenomas serosos | TNE pancreáticos | |
| Frecuencia | 70-72% | 4% | 5-17% |
| clínica | Asintomáticos o síntomas compresivos | Asintomáticos o síntomas compresivos | Crecimiento lento, asintomáticos, no funcionantes |
| Radiología | Múltiples lesiones quísticas que no realzan con contraste.Calcificaciones lineales y periféricas | Racimos de quistes < 2cm.Calcificaciones centrales que realzan con contraste «cicatriz estrellada» | Masas pancreáticas hipervasculares con cambios necróticos y realce heterogéneo |
| Prueba de imagen | TC/RM | TC/RM | TC/RM |
| Pronóstico | Benigno | Benigno | Capacidad de malignización |
RM: resonancia magnética; TC: tomografía computarizada; TNE: tumores neuroendocrinos.
Aunque menos frecuentes que los quistes, también se ha descrito la aparición de TNE en estos pacientes. El 5-10% de los tumores neuroendocrinos gastroenteropancreáticos tienen un componente hereditario conocido3. Existen varios síndromes familiares hereditarios en los que se describen este tipo de tumores como son el MEN 1, donde supone la primera causa de muerte, la neurofibromatosis tipo 1 en la que se describe de forma extremadamente rara o el VHL donde aparecen en el 5-17% y tienen un crecimiento lento.
Los TNE asociados a VHL suelen ser tumores productores de hormonas pero clínicamente no funcionantes y con mayor frecuencia son malignos. Se diagnostican al realizar pruebas de imagen ante la existencia de síntomas inespecíficos relacionados con la compresión como dolor crónico, ictericia o episodios de pancreatitis, indistinguibles de los de otras lesiones, o por la aparición de metástasis. En la TC o RM aparecen como lesiones sólidas hipercaptantes en fase arterial, encapsuladas y bien circunscritas, con realce heterogéneo y que pueden contener cambios necróticos en su interior.
El diagnóstico diferencial de los distintos tipos de lesiones pancreáticas asociadas a VHL es muy importante, ya que puede marcar el pronóstico del paciente. Sus características se resumen en la tabla 2. Es importante resaltar la baja incidencia de muerte por metástasis de tumor neuroendocrino de páncreas que Blansfield cifra en 0,31%7 y que hace más excepcional nuestro caso.
El diagnóstico de malignidad se basa en la observación de invasión vascular, de otros órganos o la presencia de metástasis linfáticas o a distancia. Varios autores12 han descrito factores asociados a malignidad en pacientes con imágenes sospechosas de TNE: tumores mayores de 3cm, mutación del exón 3 del gen VHL y el tiempo de duplicación tumoral menor de 500 días. Los pacientes con 2 de los 3 criterios deberían ser intervenidos por el alto riesgo de desarrollar metástasis hepáticas. Por otro lado, Libutti13 establece, a su vez, una serie de criterios quirúrgicos para los TNE pancreáticos en VHL: que no haya evidencia de metástasis, tumor mayor de 3cm en cuerpo o cola o mayor de 2cm en cabeza y aquellos pacientes que se someten a una laparotomía por otras lesiones. En estos casos, se debe intentar una resección económica, realizando enucleación de las lesiones siempre que sea posible. Aun así, en numerosas ocasiones la asociación de numerosos quistes en toda la glándula y la sospecha de malignidad obliga a la radicalidad en las resecciones.
Como conclusión, el seguimiento de las lesiones pancreáticas en los enfermos con enfermedad de VHL adquiere gran importancia ya que puede condicionar el pronóstico de los pacientes.

