




La pancreatitis aguda recurrente (PAR) es una entidad infrecuente en la infancia y en la adolescencia, catalogándose como idiopáticos un alto porcentaje de los casos. Los avances en el estudio genético han permitido identificar mutaciones en determinados genes que predisponen al desarrollo de la PAR.
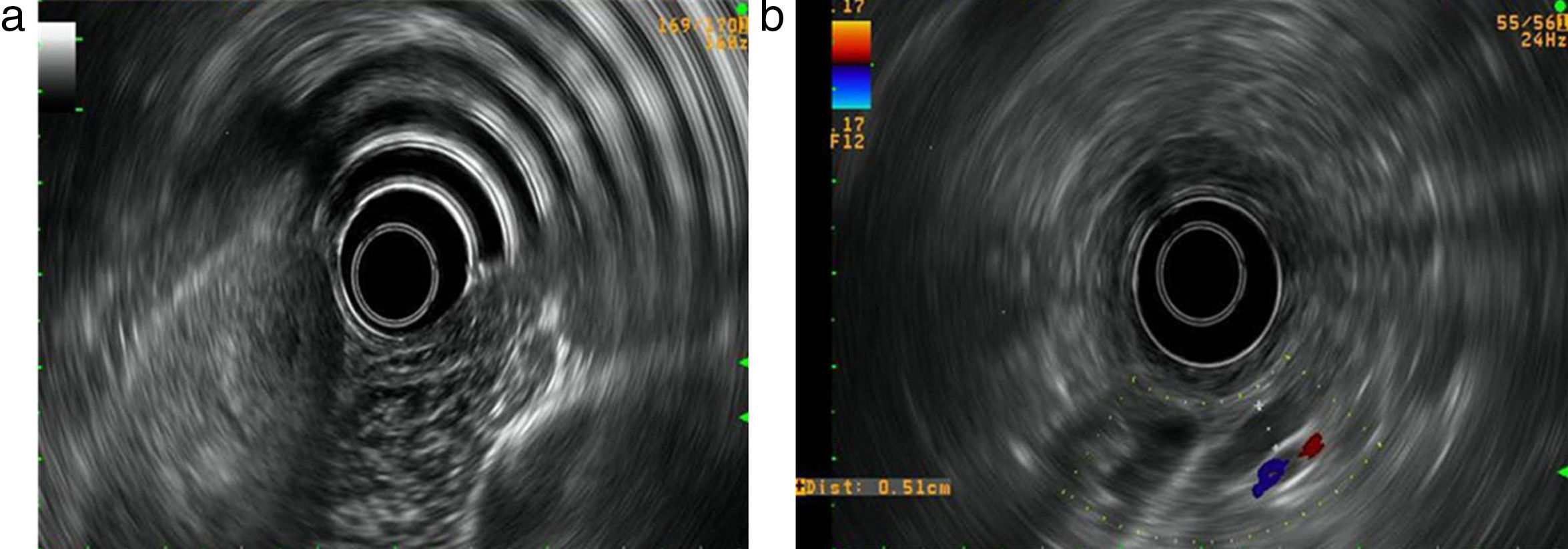
Varón de 16 años estudiado en el servicio de aparato digestivo por PAR. Desde los 14 años presenta entre 1 y 4 episodios/año, con elevación de la amilasa sanguínea (700-1.500 U/l), perfil hepático normal y sin evidencia de enfermedad biliopancreática en la ecografía abdominal. No refiere hábitos tóxicos, ni antecedentes médicos personales o familiares de interés. En su último ingreso el estudio con ecografía abdominal y resonancia magnética (RM) más colangiorresonancia (CRMN) muestra un páncreas de ecoestructura hiperecoica en cabeza y cola, dilatación del conducto pancreático principal, compatible con PC, siendo la vesícula y las vías biliares normales. Tras la resolución del episodio agudo el estudio con ecoendoscopia (USE) confirma la existencia de una PC con calcificaciones (fig. 1a y b). El resto del estudio etiológico fue normal: analítica con perfil lipídico y tiroideo, anticuerpos antinucleares, antimúsculo liso, antimicrosomales hepáticos, antimitocondriales, antilactoferrina, ceruloplasmina, factor reumatoide, inmunoglobulinas (incluida IgG4), y serología viral y de Micoplasma y Salmonella. El paciente realiza tratamiento empírico con 40mg de prednisona, volviendo a presentar 2 nuevos episodios durante su administración, por lo que fue suspendido. Finalmente se investiga el gen CFTR que revela la presencia del alelo 7T del gen en homocigosis, completando el estudio de la FQ con el test del sudor que resulta bordeline (40 mEq/l), junto con un espermiograma y estudio funcional respiratorio normales.
 Cabeza pancreática aumentada de tamaño, de ecogenicidad heterogénea, con imágenes lineales hiperecogénicas superpuestas «en hojaldre». b) Conducto pancreático principal dilatado en todo el recorrido (diámetro de 51mm) sin litiasis en su interior.")
La prevalencia de la PAR en la infancia y la adolescencia, en torno al 10-20%, es muy inferior a la hallada en la edad adulta y sus causas difieren entre ambos grupos1. El alcohol y la colelitiasis provocan la mayoría de PAR y/o PC en adultos, mientras que en menores de 18 años predominan las alteraciones anatómicas, traumatismos, trastornos metabólicos y toxicidad farmacológica2. En nuestro paciente, tras un estudio etiológico completo, el único factor predisponente para la PAR identificado fue la homocigosis para el alelo 7T del gen CFTR. El estudio autoinmune resultó negativo, y aunque existe una forma de PAI IgG4 negativa (PAI tipo 2), la ausencia de respuesta a los corticoides hicieron poco probable su diagnóstico.
Hasta un 40% de las PAR en la infancia son catalogadas de idiopáticas1. Sin embargo, las investigaciones recientes apuntan a una causa genética en un alto porcentaje de estas1,3. En una serie de 78 pacientes en edad pediátrica, el 42,3% presentaban una predisposición genética a PAR y/o una historia familiar positiva o prueba del sudor positivo o en valores bordeline4.
El gen CFTR codifica la proteína reguladora de la conductabilidad transmembrana situada en la membrana apical de las células epiteliales secretoras y absortivas de distintos órganos, y su alteración es responsable de las manifestaciones de la fibrosis quística (FQ). Las mutaciones del CFTR determinan una menor fluidez de las secreciones pancreáticas y una reducción en la secreción de bicarbonato, provocando la activación de la cascada enzimática y el daño pancreático5,6. Se han identificado unas 1.800 mutaciones con diferencias en su expresión y función proteica, desconociendo aún el significado del 40%. El nuevo sistema de clasificación de las mutaciones del gen CFTR (score de prevalencia de insuficiencia pancreática [PIP]) cataloga las mutaciones en leves (≤ 0,25) confiriendo cierta función residual del gen, o severas (> 0,25) con consecuencias funcionales más graves7. En nuestro paciente, la única alteración genética detectada fue la homocigosis para el alelo 7T del gen CFTR, previamente descrita en heterocigosis con el alelo 5T en 3 pacientes con PAR, pero no en homocigosis6.
La pancreatitis sintomática es poco común en la FQ, ocurriendo exclusivamente en pacientes con suficiencia pancreática. El riesgo de desarrollar una pancreatitis asociada a las mutaciones del gen CFTR varía con el genotipo: así los sujetos homocigotos para una mutación leve (leve/leve) son los de mayor riesgo, seguidos de los heterocigotos portadores de una mutación leve (leve/severa), frente a los homocigotos para una mutación severa (severa/severa)7,8. La evaluación de los pacientes con mutaciones del gen CFTR incluye la prueba del sudor y el potencial diferencial nasal. Los criterios diagnósticos de FQ se cumplen en el 21% de los casos, presentando el resto grados intermedios de disfunción, incluyéndose en el grupo de los trastornos relacionados con el gen CFTR5. En nuestro caso, la prueba del sudor arrojó valores en rangos bordeline, como en la mayoría de estos pacientes. No todos los portadores de mutaciones leves con suficiencia pancreática desarrollan una pancreatitis, reconociéndose la influencia de otros factores genéticos o medioambientales determinantes de la heterogeneidad de la enfermedad final en pacientes con FQ9.
Por todo lo expuesto, se recomienda el cribado genético del gen CFTR en el estudio de las PAR idiopáticas que acontece durante la infancia o en la edad adulta temprana. Se requieren más estudios para conocer el mecanismo molecular, la correlación entre el genotipo y el fenotipo, así como las interacciones con factores genéticos y medioambientales del gen CFTR.

