




La infección por el virus de la hepatitis C (VHC) representa un problema grave de salud en el mundo occidental, donde las tasas de infectados crónicamente oscilan en la mayoría de los países entre el 1,5 y el 3%. El tratamiento actual de la infección con la combinación de interferón pegilado (IFN-P) y ribavirina (RIB) consigue curar cerca del 50% de los enfermos infectados por el genotipo 1, que es de largo la forma más común (75% de los infectados).
La introducción de un tratamiento triple que, además de IFN-P y RIB, incluye un agente antiviral directo (AAD) consistente en una antiproteasa frente a la proteasa NS3/4 del VHC, como el telaprevir o el boceprevir, permitirá obtener una tasa de curación del 75% de los enfermos que no habían recibido nunca ningún tratamiento (enfermos naïve) y del 50% en los que había fracasado un tratamiento previo. Los AAD no están actualmente indicados en el tratamiento de la hepatitis crónica C causada por otros genotipos.
El tratamiento triple tiene una eficacia superior a la del tratamiento clásico (IFN-P+RIB), pero ocasiona más efectos secundarios y, además, implica la necesidad de una evaluación más entretenida de los enfermos que se tienen que tratar y un seguimiento más frecuente. Estos hechos pueden tener un impacto muy negativo en los centros que atienden enfermos con hepatitis C, puesto que se pueden ver sometidos a una presión asistencial muy superior a la actual, ya que tendrán que atender los enfermos con una hepatitis crónica C previamente no tratada que se vayan diagnosticando, que tendrán que ser evaluados por si tienen que recibir tratamiento triple, y también muchos de los enfermos en los que ha fracasado un tratamiento previo con IFN-P y RBV y siguen siendo visitados en las consultas de hepatitis de todos los hospitales. Finalmente, se tendrá que añadir a los enfermos que tendremos que tratar, aquellos que prefirieron esperar a tratarse de la hepatitis C a que dispusiéramos de un tratamiento más efectivo.
La incorporación de AAD incrementará la eficacia del tratamiento y acortará en muchos enfermos la duración del tratamiento, pero también aumentará notablemente el coste, el número y la intensidad de sus efectos adversos. Por consecuente, parece razonable hacer una reflexión sobre cuál sería la forma que permitiría la máxima eficiencia (mejores resultados posibles con el coste de la acción que se emprenda) y la mayor seguridad de los pacientes (prevención de los efectos adversos y actuación lo más rápida posible cuando aparecen). Es responsabilidad de la comunidad médica establecer las normas y fijar las recomendaciones, pero seguirlas es una obligación ética de los médicos que tratan enfermos.
La máxima eficiencia se conseguirá aplicando las siguientes medidas:
- 1)
Prescribir el tratamiento convencional (IFN-P y RIB), sin añadir AAD, a los enfermos que tienen muchas posibilidades de curarse con el tratamiento doble.
- 2)
Prescribir el tratamiento triple a los enfermos que seguramente no presentarían una respuesta viral sostenida con tratamiento convencional.
La máxima seguridad puede conseguirse de la siguiente manera:
- 1)
Cumpliendo con las indicaciones de la ficha técnica en cuanto a las contraindicaciones absolutas y relativas de los AAD, y de los criterios de suspensión del tratamiento. Especial relevancia tiene la valoración de interacciones potenciales con otros fármacos que pueden contraindicar el tratamiento o exigir ajustar las dosis.
- 2)
Siendo el médico asequible al enfermo que está en tratamiento y que presente algunos de los efectos no deseados del mismo, el manejo de los cuales no pueda esperar a la visita planificada más inmediata. Esta asequibilidad implica la posibilidad de consulta con especialistas que puedan abordar el manejo de problemas extrahepáticos, como depresión o manifestaciones cutáneas.
El tratamiento triple plantea incógnitas que todavía no podemos resolver. En primer lugar ignoramos si se dispondrá de suficientes recursos para afrontar las demandas de tratamiento, recursos tanto económicos, como de personal médico dedicado a estos enfermos. Sin embargo, se tendrán que aportar los argumentos necesarios para evitar unas excesivas limitaciones, sobre todo porque la curación de los enfermos que podrían desarrollar una cirrosis si no se trataran tiene que representar a la larga un ahorro en atención médica, incluyendo en trasplantes, así como muchos sufrimientos físicos y morales. Los médicos hepatólogos tendrán que ser muy cuidadosos al hacer indicaciones cuidadosas del tratamiento, valorando la motivación de los enfermos, que los haga tolerantes a los efectos adversos causados por la medicación, las posibilidades de respuesta, así como el riesgo que tenga la enfermedad de empeorar. También tenemos que tener en cuenta que la mayor sobrecarga de trabajo que comportará este nuevo tratamiento para los centros de excelencia tendrá que ser tenido en cuenta por los equipos directivos de las instituciones para no reducir la dimensión de los equipos encargados de estos tratamientos o idealmente para reforzarlos.
También sería conveniente hacer un esfuerzo para calcular cuál deberá ser el tiempo necesario para atender una primera visita, en la que se tiene que hacer la evaluación inicial, y cuánto tiempo hará falta para las visitas sucesivas. Esta información sería muy útil para hacer una previsión de cómo tendremos que afrontar de manera global este reto sanitario.
Evaluación clínica de los enfermos con hepatitis crónica c para decidir si se tienen que tratar y qué tratamiento tienen que recibirLa buena praxis médica justifica el seguimiento de la sistemática siguiente en los enfermos con hepatitis crónica C en los que se prevé la aplicación de un tratamiento antiviral:
- 1)
Determinación del genotipo del virus y de la carga viral para poder establecer la estrategia terapéutica más conveniente.
- 2)
Estimación del grado de fibrosis hepática del paciente a través de cualquier de los siguientes procedimientos:
- a)
Biopsia hepática reciente, aplicando un sistema de puntuación de la fibrosis como METAVIR1, los criterios de Scheuer2,3 u otro.
- b)
Fibroscan, considerando que hay una fibrosis significativa (equivalente a F2 de la clasificación METAVIR o más) a partir de un valor de 7,6 kPa4,5.
- c)
Alguno de los métodos bioquímicos indirectos de uso común6–8.
- a)
- 3)
Determinar el polimorfismo de la IL28B para saber si el paciente presenta el genotipo favorable (el CC) que se asocia a una mayor respuesta al tratamiento con IFN-P+RIB, u otro no favorable (los genotipos CT y TT)9,10.
- 4)
Clasificar los enfermos con hepatitis crónica C en alguna de las siguientes categorías según si habían recibido o no un tratamiento previo con IFN-P y RIB y, en caso afirmativo, cuál fue el resultado:
- a)
Enfermos previamente no tratados o naïve.
- b)
Recidivantes después de un tratamiento previo con IFN-P+RIB.
- c)
No respondedores con respuesta parcial a IFN-P+RIB.
- d)
No respondedores por tratamiento inadecuado o en quienes se ignora cómo se comportó el enfermo durante el tratamiento.
- e)
No respondedores absolutos (null responders) a IFN-P+RIB.
- a)
- 1.
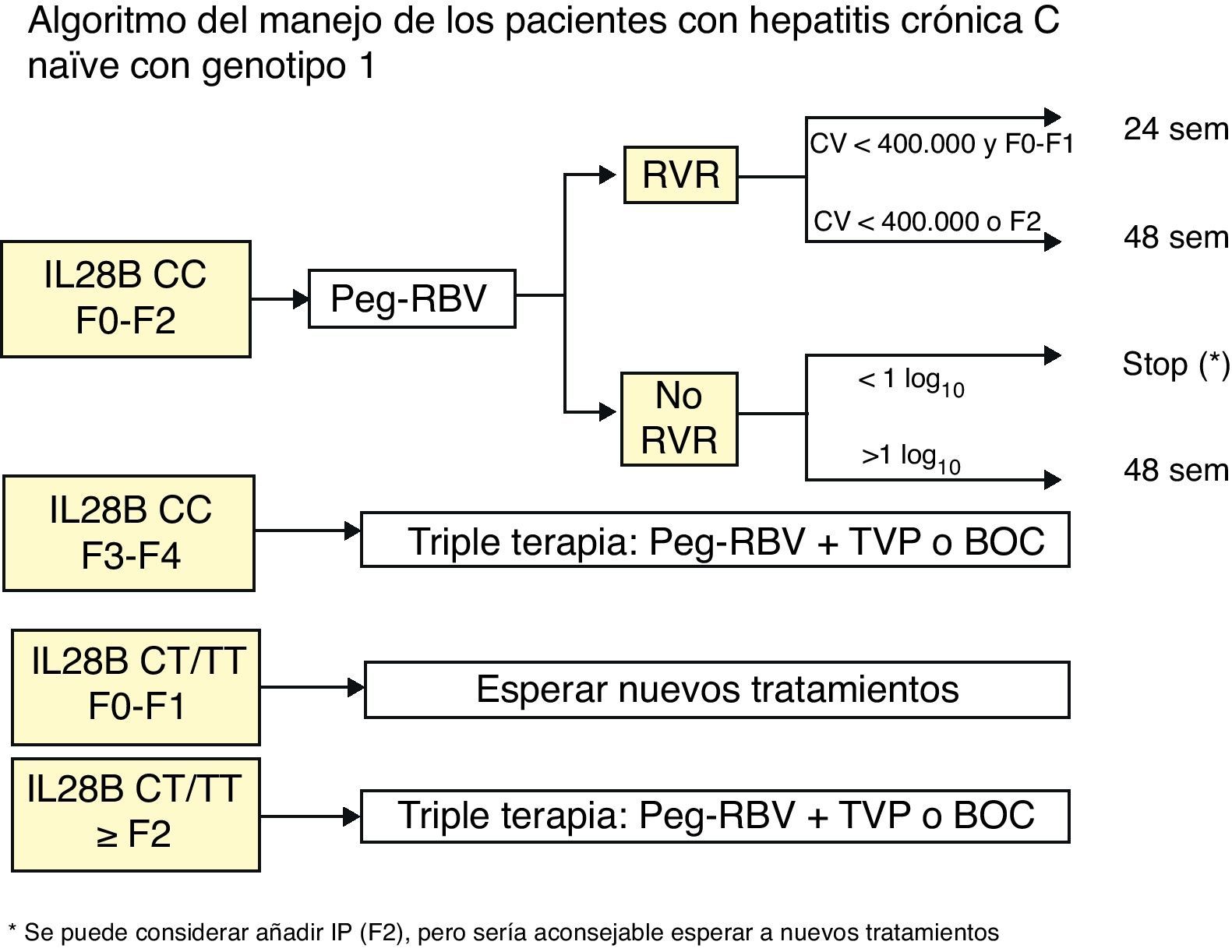
Enfermos con genotipo CC de la IL28B y fibrosis<F3. Se recomienda el tratamiento con IFN-P y RIB:
- a)
Si hay una respuesta viral rápida (RVR=RNA-VHC negativo a las 4 semanas de tratamiento) conviene seguir con el tratamiento doble hasta cumplirse 24 o 48 semanas de tratamiento. Si el enfermo no tiene fibrosis (F0) o ésta es mínima (F1) se puede mantener el tratamiento doble hasta 24 semanas cuando la carga viral basal es inferior a 400.000 U/ml; en el resto de casos se debe mantener hasta 48 semanas.
- b)
Si no hay RVR pero la carga viral ha caído>1 log10 hay que mantener el tratamiento hasta 48 semanas. Si la carga viral ha caído<1 log10 lo más aconsejable es parar el tratamiento a la espera de nuevas terapias.
- a)
- 2.
Enfermos con genotipo CC de la IL28B y fibrosis F3 o F4. Iniciar tratamiento triple, con 4 semanas previas de tratamiento con IFN-P+RIB (lead-in) si se quiere dar boceprevir, tal como indica la ficha técnica, o directamente sin lead-in si se utiliza telaprevir. Si los enfermos se tratan con boceprevir y no se detecta el RNA-VHC a las semanas 8 y 24, el tratamiento debe tener una duración de 28 semanas (4 semanas de IFN-P+RIB seguidas de 24 semanas de tratamiento triple, es decir, IFN-P, RIB y boceprevir). Si el paciente presenta una cirrosis el tratamiento será de 48 semanas. Si los enfermos se tratan con telaprevir y no se detecta el RNA-VHC a las semanas 4 y 12, la duración del tratamiento será de 24 semanas (12 semanas de terapia triple, incluyendo telaprevir, seguidas de 12 semanas de terapia doble con IFN-P+RIB). Si todavía se detecta RNA-VHC a la semana 12 o si el paciente presenta una cirrosis el tratamiento se tendrá que mantener 48 semanas.
- 3.
Enfermos con genotipos TT o CT del IL28B y fibrosis F2, F3 o F4. Iniciar tratamiento triple, como se indica en el apartado anterior.
- 4.
Enfermos con genotipos TT o CT del IL28B y sin fibrosis (F0) o fibrosis F1. Se recomienda esperar a tratamientos mejores.
Tratarlos todos con tratamiento triple siguiendo las indicaciones de ficha técnica de boceprevir y telaprevir. Si el paciente se trata con boceprevir la duración del tratamiento será de 48 semanas (4 semanas de IFN-P+RIB, seguidas de 32 semanas de terapia triple con boceprevir más IFN-P+RIB y 12 semanas de IFN-P+RIB si no hay cirrosis); en el caso de que el enfermo tenga una cirrosis es aconsejable que el tratamiento triple sea de 44 semanas, pero podría ser de 32 semanas seguidas de 12 semanas de IFN-P+RIB en caso de intolerancia). Si el paciente se trata con telaprevir y el RNA-VHC no es detectable a las semanas 4 y 12 la duración del tratamiento será de 24 semanas (12 semanas de terapia triple, seguidas de 12 semanas de IFN-P+RBV). Si se detecta RNA-VHC o si el paciente tiene una cirrosis el tratamiento será de 48 semanas (12 semanas de terapia triple, seguidas de 36 semanas de IFN-P+RBV).
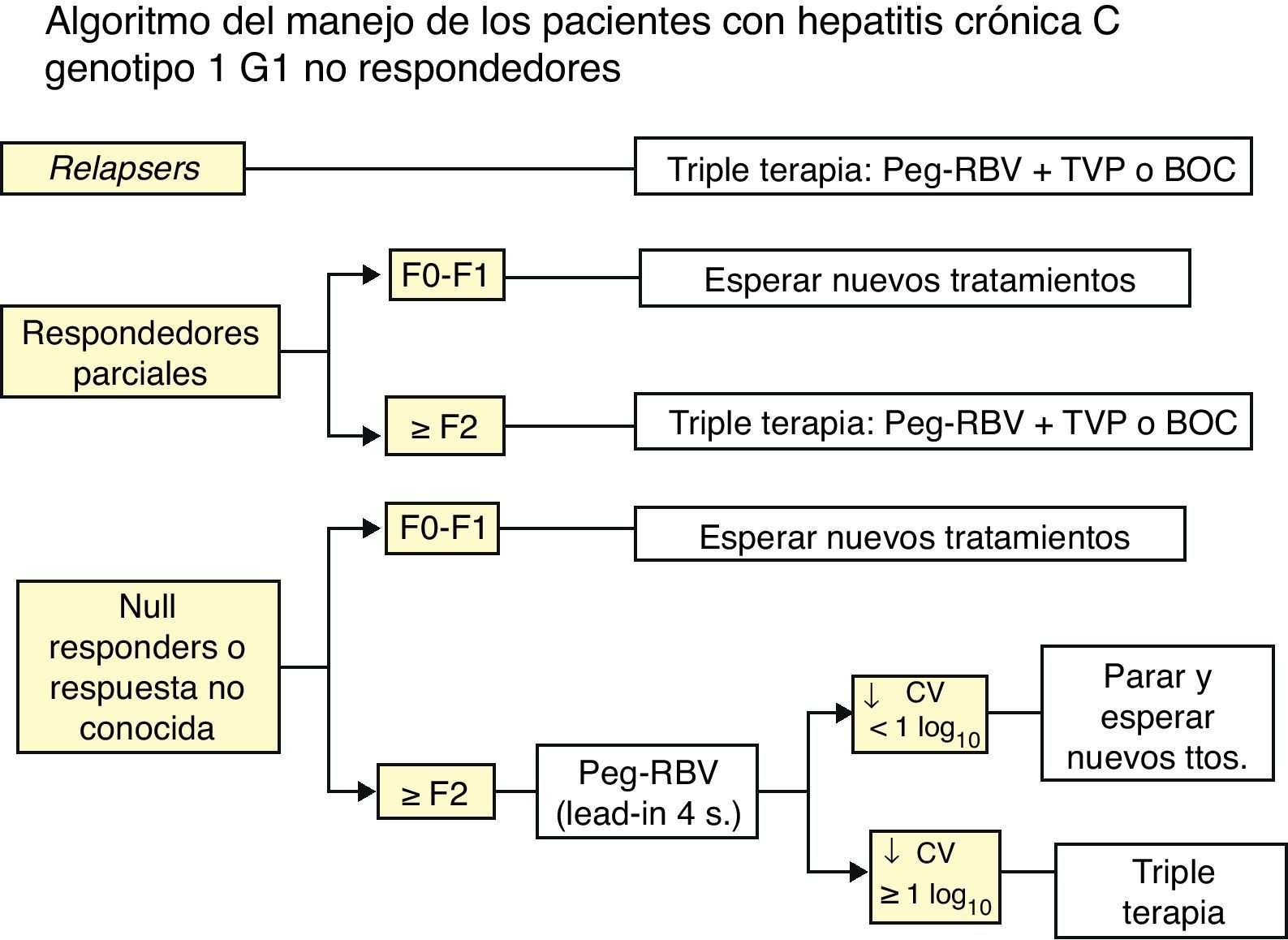
Enfermos no respondedores con respuesta parcial a un tratamiento previo con IFN-P y RIBLa decisión se tomará según si hay o no fibrosis, mediante Fibroscan o biopsia hepática o métodos indirectos:
a) Si no hay fibrosis (F0) o esta es mínima (F1) se recomienda esperar a tratamientos mejores.
b) Si la fibrosis es significativa (≥ F2) se recomienda tratamiento triple, siguiendo ficha técnica.
Enfermos con respuesta nula a un tratamiento previo con IFN-P y RIB y en los que se ignora si este fue inadecuado o hubo una respuesta parcialSe aconseja hacer en todos los enfermos un tratamiento inicial de 4 semanas con IFN-P+RIB (lead-in) y decidir la continuidad del tratamiento según haya o no respondida.
- a)
Si ha habido respuesta (caída de>1 log10 del RNA del VHC).añadir un AAD (tratamiento triple) y mantener el tratamiento durante 48 semanas.
- b)
Si no ha habido respuesta, es decir, no caída del RNA del VHC de al menos 1 log10 a las 4 semanas, se recomienda parar el tratamiento.
Un esquema resumido del tratamiento de los enfermos naïve y de los no respondedores se presenta en las figuras 1 y 2.


Telaprevir: 750mg (2 comprimidos) cada 8 h, administrados con comida.
Boceprevir: 800mg (4 cápsulas) cada 8 h, administrados con comida. Los comprimidos se tienen que tragar enteros sin masticar ni disolver.
Regla de suspensiónEn los pacientes tratados con telaprevir si el RNA-VHC es>1.000 UI/ml a la semana 4 o a la semana 12 después del inicio del tratamiento, los 3 fármacos tienen que ser suspendidos. En los pacientes tratados con boceprevir si el RNA-VHC es>100 U/ml a la semana 12 después del inicio del tratamiento triple, los 3 fármacos tienen que ser suspendidos. Si el RNA-VHC a la semana 24 del inicio del tratamiento triple es>25 U/ml parar el tratamiento.
Exclusiones de la indicación de un tratamiento triple por la hepatitis crónica C- 1.
Se excluirán de la indicación de tratamiento triple los enfermos con hepatitis crónica C de grupos especiales en los que todavía no hay bastantes estudios que justifiquen su uso o en los que todavía no está claro si puede haber interacciones de los AAD con los fármacos que los enfermos reciben como tratamiento de la enfermedad de base: a) cirróticos descompensados; b) enfermos en hemodiálisis; c) enfermos pediátricos; d) coinfectados con VIH; e) trasplantados hepáticos, y f) drogadictas en tratamiento sustitutivo con metadona (por desconocimiento de si hay interacciones con AAD). Estos subgrupos de pacientes podrán ser tratados cuando se disponga de más información sobre interacciones y eficacia del tratamiento.
- 2.
Tampoco habrá indicación de tratamiento triple si el médico tiene dudas razonables de que habrá un buen cumplimiento.
- 3.
Finalmente tampoco se tendrían que tratar los enfermos que no estén claramente motivados y decididos a asumir los efectos adversos si se producen. A tal efecto los enfermos tendrán que llenar un documento de consentimiento informado antes de empezar (anexo 1).
Como que se trata de una enfermedad bastante común, en la mayoría de los centros hospitalarios de Cataluña hay especialistas que han adquirido el entrenamiento necesario para tratar la hepatitis crónica C con IFN-P y RIB, y para actuar cuando aparecen efectos adversos. La incorporación de un AAD, como telaprevir o boceprevir, al tratamiento convencional plantea problemas nuevos, como es el manejo de los efectos adversos inducidos por los AAD, y el acceso rápido a los resultados de las pruebas de laboratorio necesarias para actuar con racionalidad y eficiencia. Prolongar indebidamente el tratamiento con los AAD, cuando no se produce respuesta, incrementa la aparición de mutaciones del virus que determina la formación de cepas resistentes, e incrementa innecesariamente el coste. Conviene, por lo tanto, que los médicos dispuestos a aplicar tratamientos triples trabajen en centros que reúnan las siguientes características:
- a)
Tener acceso a los resultados de la determinación cuantitativa del RNA del VHC en un tiempo no superior a una semana.
- b)
Igualmente, tienen que poder disponer de los resultados del examen de los polimorfismos del IL28, hecho en el laboratorio del propio hospital o en un laboratorio externo que pueda hacer esta determinación de manera regular y rápida.
- c)
Disponibilidad de un dermatólogo para atender consultas relacionadas con lesiones cutáneas aparecidas durante el tratamiento.
- d)
Accesibilidad fácil por consultas urgentes con psiquiatra experto en efectos secundarios de carácter psiquiátrico relacionados con el IFN-P, previas o durante el tratamiento. Los médicos que no puedan reunir estos condicionantes tendrían que poder remitir los enfermos que consideren potencialmente tributarios de este tratamiento a otro centro. Seguramente no sería necesario efectuar acreditaciones formales de los centros hospitalarios que puedan indicar y dispensar el tratamiento triple para conseguir la máxima rentabilidad del uso de los agentes antivirales directos y asegurar un control adecuado de los efectos adversos relacionados con la medicación. El buen criterio ético de los médicos especialistas tendría que permitir que los tratamientos triples se hicieran solo en lugares donde dispongan de los condicionantes señalados y que no los aplicaran médicos que no estén suficientemente familiarizados con los efectos adversos causados por estos fármacos y las posibles interacciones con otros medicamentos que pueda recibir el enfermo (anexo 2).
Es recomendable que antes de iniciarse un tratamiento triple el médico obtenga un documento de consentimiento informado, debidamente firmado por el paciente, después de darle la información sobre los beneficios e inconvenientes de este tratamiento (anexo 1).
¿Qué hacer con los enfermos en los que no se indica el tratamiento o este ha sido ineficaz?Para estos enfermos no tenemos en el momento actual ningún tratamiento. Hay cerca de 30 moléculas en estudio, en fase II e III, que en un plazo de muy pocos años nos permitirán disponer de recursos más potentes y con menos efectos adversos que los actuales.
Conflicto de interesesR. Solà declara haber recibido honorarios por consultorías de Gilead Sciences, Roche/Genentech, Roche Pharma, Tibotec, Jansen Cilag, Schering-Plough/Merck y Bristol Myers Squibb; y por ponencias de Bristol Myers Squibb, Gilead Sciences, Novartis, Roche/Genentech, Roche Pharma, y Merck Sharp & Dome; R. Planas ha recibido honorarios de Roche, MSD, Janssen, Gilead, BMS, y Boehringer Ingelheim; X. Forns ha recibido Becas de investigación de Roche y MSD y Honorarios por consultoría de Janssen y MSD. R. Esteban ha recibido honorarios de MSD, Janssen, Glaxo, Gilead, BMS y Novartis.
Los Dres. M. Bruguera, J.A. Quer y M. Vergara no reconocen conflicto de intereses.
Telaprevir. Telaprevir puede causar diferentes efectos adversos (EA) que se añadirán a los ya conocidos del interferón (IFN) y la ribavirina (RIB)1–3. En el estudio ADVANCE se suspendió el tratamiento por EA en un 10% de los pacientes que recibieron telaprevir durante 12 semanas, con IFN pegilado (IFN-P) y RIB durante 48 semanas, hecho que solamente se produjo en el 7% de los que recibieron la terapia estándar. Estos valores fueron del 15 y el 3%, respectivamente, en el estudio REALIZE1,3.
Entre los EA destacan los de tipos dermatológico. El más frecuente el exantema cutáneo, reportado en un 56% de los pacientes que reciben telaprevir. Presentaron prurito un 47% de los casos tratados. Hay que distinguir los EA cutáneos de las lesiones debidas a la propia infección por el VHC, como el liquen plano, la porfiria cutánea tarda o las debidas a la crioglobulinemia mixta8. También tendremos que diferenciar las lesiones cutáneas atribuibles al tratamiento con IFN9, como el exantema leve o moderado, que llega a grave en un 5% de los pacientes. Un dato importante de los estudios publicados es que solamente fue necesario suspender el telaprevir para controlar el exantema en un 6% de casos, y que en la mayoría de ellos se pudo proseguir el tratamiento con IFN-P y RIB. Solo en el 1% de los pacientes se interrumpió por completo el tratamiento debido a la gravedad de las lesiones cutáneas. Habitualmente el exantema mejora y desaparece después de la finalización o la interrupción del tratamiento con telaprevir, aunque en algún caso puede tardar semanas a resolverse.
Se trata de una erupción de tipo eccematoso y maculopapuloso que suele aparecer durante las primeras 4 semanas del tratamiento (media de 25 días), aunque puede hacerlo en cualquier momento de la terapia. De forma excepcional (< 1%) se han descrito casos de síndrome de Stevens-Johnson, síndrome de exantema farmacológico con eosinofilia y síntomas sistémicos (DRESS), pustulosis exantemática generalizada aguda (PEGA), eritema multiforme (EM) y necrólisis epidérmica tóxica (NET)10,11. En el estudio ADVANCE se desarrolló un protocolo de gradación del exantema cutáneo secundario al uso de telaprevir que, con algunas modificaciones, ha permitido establecer una serie de pautas y recomendaciones para su manejo práctico1,12:
Grado 1, leve. Erupción cutánea localizada en una o varias zonas del cuerpo, con distribución limitada, con o sin prurito. Es una erupción leve y superficial, que nunca está asociada a signos de disrupción de la epidermis o afectación de las mucosas.
Grado 2, moderado. Definido como una erupción cutánea con afectación difusa pero siempre inferior al 50% de la superficie corporal, que puede acompañarse de separación de la piel superficial y de signos inflamatorios en las mucosas. Estos tendrán que diferenciarse de aquellas lesiones mucosas específicas no necesariamente relacionadas con reacciones cutáneas, como la estomatitis, las aftas bucales o el liquen plano. En esta fase puede haber síntomas y signos sistémicos de intensidad moderada, como fiebre, artralgias o eosinofília.
Grado 3, grave. Exantemas generalizados con afectación de más del 50% de la superficie corporal, con lesiones de tipos vesicular o ampolloso, úlceras superficiales de las mucosas, desprendimiento epidérmico con separación de la dermis subyacente, púrpura palpable o eritema que no desaparece a la presión. También puede haber en este grado firmas y síntomas sistémicos asociados a una PEGA, una EM o a un DRESS. Este solo suele presentarse entre las semanas 5 y 10 de tratamiento, en forma de fiebre elevada, edema facial, adenopatías, eosinofilia y aparición de linfocitos atípicos, disfunción renal (elevación de creatinina) y alteraciones hepáticas (hepatomegalia, elevación de transaminasas y fosfatasas alcalinas), coincidiendo con una rápida progresión del exantema13,14. La aparición de una erupción ampollosa generalizada, el síndrome de Stevens-Johnson o la necrólisis epidérmica tóxica tienen que ser consideradas como complicaciones graves y potencialmente mortales en estos pacientes. Todas ellas tienen que ser detectadas el más precozmente posible10,11.
Todos los pacientes con lesiones cutáneas atribuibles al tratamiento tienen que ser vigilados de forma estrecha, con una periodicidad variable que dependerá del grado de lesión. Los pacientes con lesiones leves (grado 1) pueden ser tratados con corticosteroides tópicos del tipo de la clobetasona y la triamcinolona no más de 2 semanas. Se utilizarán preferentemente en forma de cremas o lociones, puesto que las pomadas y los hielos tienen un menor potencial de absorción. Conviene evitar el uso de corticosteroides sistémicos, dado su potencial efecto inmunosupresor, que podría repercutir en el comportamiento de la infección vírica y presentar posibles interacciones farmacológicas con los AAD.
En esta fase puede ser de utilidad asociar antihistamínicos tópicos y sistémicos, como difenhidramina, levocetiricina, hidroxicina o desloratadina, que producirán un alivio sintomático del picor. Conviene evitar la exposición al sol. En general, la existencia de lesiones leves no requiere la modificación del tratamiento. Cuando las lesiones son más importantes (grado 2) o se produce una progresión de las lesiones leves, se mantendrán las mismas medidas, pero la no mejora o evolución de la exantema obligará a plantear la interrupción del telaprevir. Si a pesar de esta maniobra las lesiones no mejoran, después de un plazo de 7 días se considerará la interrupción temporal de la RIB, puesto que en ocasiones este fármaco podría ser la causa del exantema. La terapia con IFN-P podrá mantenerse, puesto que es muy improbable que este fármaco se relacione con las lesiones dérmicas.
La existencia de lesiones graves (grado 3) exige la interrupción inmediata del telaprevir. También ante un síndrome de Stevens-Johnson, DRESS, PEGA o EM, se tiene que interrumpir de forma inmediata y definitiva toda la terapia. En cualquiera de las situaciones descritas que impliquen la interrupción de telaprevir por un EA, esta será definitiva. No se debe retomar el fármaco aunque desaparezcan las lesiones. El exantema suele desaparecer entre 7 y 14 días desde que se suspende o finaliza el tratamiento. El segundo EA remarcable relacionado con el uso de telaprevir es la anemia, definida por unos valores de hemoglobina (Hb) inferiores a 10g/dl, que se ha reportado en el 36% de los pacientes en los estudios de registro (17% en los cuales reciben la terapia estándar). La incidencia de anemia grave (Hb < 8,5g/dl) fue del 14%, en comparación al 5% que presentaban los pacientes tratados con IFN-P y RIB1–3.
Hasta un tercio de los pacientes requirieron la reducción o la interrupción del tratamiento con RIB para controlar la anemia. Se consideró necesario suprimir el telaprevir por este motivo, solamente en un 4% de los casos. Precisaron transfusión de sangre un 12% de los pacientes tratados con telaprevir que presentaban unas cifras de hemoglobina por debajo de 10 g/dl (5% en el grupo control). En la práctica, resulta difícil distinguir si la anemia está producida por el telaprevir o la ribavirina. En los pacientes que finalizan las 12 semanas de tratamiento con telaprevir y que han presentado anemia, los valores suelen mejorar de forma gradual hasta conseguir, hacia la semana 20, cifras similares al que sucede en enfermos que reciben la terapia estándar.
El manejo inicial de la anemia tendrá que basarse siempre en la reducción de la dosis de RIB. En los estudios de registro (ADVANCE, ILLUMINATE)1,2 se ha analizado la repercusión de la modificación de la dosis de ribavirina sobre las tasas de respuesta virológica sostenida (RVS), sin observarse diferencias entre los pacientes que recibieron reducción de dosis (RVS del 76%) y los que mantuvieron la dosis completa (72%). Tampoco se apreciaron diferencias al analizar la RVS en relación con los valores de hemoglobina15. Una norma fundamental a tener en cuenta es que en aquellos pacientes en que se decida la suspensión definitiva de la ribavirina, tendremos que proceder a la interrupción del tratamiento con telaprevir de forma permanente.
Los agentes estimuladores de la eritropoyesis (AEE) podrían tener un papel en el manejo de las anemias muy sintomáticas, pero en los estudios de registro del telaprevir no se consideró está medida terapéutica y, por lo tanto, no existen datos publicados sobre su uso y eficacia. Si se considerara esta posibilidad, los AEE tendrán que utilizarse en función de la práctica clínica habitual y siguiendo las recomendaciones y precauciones que marca la ficha técnica del producto. Otros efectos adversos relevantes son de tipo digestivo, como náuseas (39%), diarrea (26%), vómitos (13%) o disgeusia (10%). En un tercio de los pacientes que reciben telaprevir se ha detectado la aparición de síntomas anorrectales, como prurito anal, hemorroides, malestar en forma de escozor y tenesmo rectal. En estos casos se puede recomendar la adición de fibra en la dieta, loperamida y tratamientos tópicos con preparados de hidrocortisona, anestésicos locales o antihistamínicos, aunque su eficacia es relativa. Estas manifestaciones raramente obligan a la interrupción del tratamiento y desaparecen después de la finalización del mismo. Es importante informar a los pacientes de su presencia y su limitación en el tiempo, remarcando que la terapia con telaprevir solamente se mantendrá durante 12 semanas.
Los pacientes que reciban tratamiento con telaprevir tendrán que ser visitados de forma periódica con controles analíticos que contengan, como mínimo, un hemograma, niveles de transaminasas, bilirrubina y creatinina séricas.
Boceprevir. En los diferentes estudios de registro, los EA más frecuentes relacionados con el uso de boceprevir fueron la anemia, la neutropenia y la disgeusia4,5. Entre todos ellos destaca, por su impacto y frecuencia de aparición, la anemia. Concretamente, de los 1.225 pacientes naïves que habían recibido este fármaco en terapia triple (SPRINT-2), el 50% desarrollaron anemia (Hb<10 g/dl), un 25% neutropenia y el 35% disgeusia, comparados con valores del 30, 19 y 16%, respectivamente, en los 467 que fueron tratados solamente con IFN-P y RIB4.
La frecuencia de aparición de anemia en los pacientes que recibieron boceprevir en el estudio RESPOND-2, que trataba pacientes no respondedores a una terapia convencional previa, fue del 45% (20% en el grupo de tratamiento estándar). En este estudio, un 44% de los pacientes que recibieron terapia triple presentaron disgeusia, que no representó ningún problema grave ni requirió modificaciones del tratamiento5. Presentaron una anemia grave (Hb<8,5mg/dl), el 6% de los pacientes naïves y el 10% de los cuales eran retratados con triple terapia (valores respectivos del 3 y el 1% con terapia estándar). En todos los estudios publicados, se paró el tratamiento en el 13% de los pacientes tratados con boceprevir, porcentaje similar al de aquellos que recibieron terapia estándar. Requirieron un ajuste de la dosis de RIB o IFN, un 39% en el grupo boceprevir y un 24% en el de terapia convencional, siendo la anemia la causa más habitual de esta medida.
Es importante comentar que en los estudios que utilizaban boceprevir, por protocolo, la anemia podía ser manejada mediante modificación de la dosis de RIB o con la administración de AEE. Es remarcable que el 43% de los pacientes de los grupos tratados con el AAD recibieron eritropoyetina para controlar la anemia (23% en el grupo de terapia estándar). Requirieron transfusión de sangre un 3% de los casos.
Un aspecto interesante a analizar es el efecto de la anemia y de sus diferentes medidas de manejo sobre la eficacia del tratamiento. En el estudio SPRINT-24, la tasa de RVS de los pacientes tratados con boceprevir que no presentaron anemia durante el tratamiento fue del 68%, mientras que en aquellos que si la presentaron y requirieron medidas para su control, como la administración de AEE, la reducción de la dosis de ribavirina o ambas, la RVS era superior al 70%16. Los pacientes que presentan anemia significativa durante el tratamiento suelen tener una concentración plasmática de ribavirina superior a la esperada para la dosis administrada, traduciendo una sobreexposición al fármaco. Cuando se reduce la dosis, la concentración plasmática suele situarse en los valores adecuados, teóricamente sin menguar su efectividad. Esta observación sugiere que la reducción de la dosis de ribavirina tendría que ser la primera medida a aplicar para el manejo de la anemia en estos pacientes.
La neutropenia se presentó en los estudios de registro con una frecuencia significativamente superior en los pacientes que recibieron boceprevir, aunque en la mayoría no requirió ningún tipo de medida. En el estudio SPRINT-2 se administraron agentes estimuladores de los granulocitos en un 10% de los pacientes de los grupos tratados con boceprevir y en el 6% del grupo de terapia estándar4
No es infrecuente que los pacientes que van a iniciar un tratamiento de la hepatitis crónica C puedan estar recibiendo o precisen otros fármacos. El conocimiento de las posibles interacciones farmacológicas de los nuevos AAD es de gran importancia, puesto que su aparición podría conducir a una disminución de eficacia del tratamiento, un incremento del riesgo de resistencias y un aumento de los EA.
Tanto telaprevir como boceprevir pueden producir una inhibición de enzimas relacionadas con el metabolismo hepático de diferentes medicamentos y de las proteínas transportadoras como la P-glucoproteína (P-gp)17,18. Se han descrito interacciones de los AAD con diferentes tipos de fármacos, como estatinas, antirretrovirales, antiarrítmicos, anticonvulsivantes, derivados ergotamínicos, tuberculostáticos y procinéticos17,18. En los pacientes trasplantados se tiene que tener en cuenta la interacción de los AAD con los fármacos inmunosupresores. Telaprevir y boceprevir incrementan de forma significativa la concentración plasmática de tacrolimus y de ciclosporina19. Además, es habitual que los pacientes trasplantados reciban otros fármacos que también utilizan el citocromo P450 por su metabolización, como sirolimus, micofenolato, macrólidos, antagonistas del calcio, estatinas y antirretrovirales.
Es fundamental revisar y anotar toda la medicación que puedan estar tomando los pacientes que van recibir una terapia triple. Antes de empezar la terapia, es recomendable consultar la ficha técnica de la AAD y alguna fuente de datos contrastada de las cuales en estos momentos son asequibles vía online (www.accesdata.fda.gov/scripts/cder/drugsatfda/index.cfm; http://222.drug-interactions.com; www.hep-druginteractions.org). Los pacientes tendrán que ser informados de la necesidad de comunicar inmediatamente a su especialista la prescripción de cualquier otro fármaco durante el período de tratamiento.
Bibliografía
1. Jacobson IM, McHutchison JG, Dusheiko GM, Di Bisceglie AM, Reddy KR, Bzowej NH, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405–16.
2. Sherman KE, Flamm SL, Afdhal NH, Nelson DR, Sulkowski MS, Everson GT, et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med. 2011;365:1014–24.
3. Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, et al. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364:241728.
4. Poordad F, McCone J, Bacon BR, Bruno S, Manns MP, Sulkowski MS, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1195–206.
5. Bacon B, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1207–17.
6. McHutchison J, Everson GT, Gordon SC, Jacobson IM, Sulkowski M, Kauffman R, et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med. 2009;360:1827–38.
7. McHutchison J, Manns MP, Muir AJ, Terrault NA, Jacobson IM, Afdhal NH, et al. Telaprevir for previously treated chronic HCV infection. N Engl J Med. 2010;362:1292–303.
8. Jacobson IM, Cacoub P, Dal Maso L, Harrison SA, Younossi ZM. Manifestations of chronic hepatitis C virus infection beyond the liver. Clin Gastroenterol Hepatol. 2010;8:1017–29.
9. Mystry N, Shapero J, Crawford RI. A review of adverse cutaneous drug reactions resulting from the use of interferon and ribavirin. Can J Gastroenterol. 2009;23:677–83.
10. Roujeau JC, Stern RS. Severe adverse cutaneous reactions to drugs. N Engl J Med. 1994;331:1272–85.
11. Roujeau JC, Allanore L, Liss Y, Mockenhaupt M. Severe cutaneous adverse reactions to drugs (SCAR): definitions, diagnostic criteria, genetic predisposition. Dermatol Sinica. 2009;27:203–9.
12. Cacoub P, Bourlière M, Lübbe J, Dupin N, Buggisch P, Dusheiko G, et al. Dermatological side effects of hepatitis c and is treatment: Patients management in the era of direct-acting antivirals. J Hepatol. 2011 [Epub ahead of print].
13. Cacoub P, Musette P, Descamps V, Meyer O, Speirs C, Finzi L, et al. The DRESS syndrome: A literature review. Am J Med. 2011;124:588–97.
14. Montaudié H, Passeron T, Cardot-Leccia N, Sebbag N, Lacour JP. Drug rash with eosinophilia and systemic symptoms due to telaprevir. Dermatology. 2010;221:303–5.
15. Sulkowski MS, Reddy R, Afdhal NH, Di Bisceglie AM, Zeuzem S, Poordad F, et al. Anemia had no effect on efficacy outcomes in treatment-naive patients who received telaprevir-based regimens in the ADVANCE and ILLUMINATE phase 3 studies. J Hepatol. 2011;54 Suppl 1:S195.
16. Sulkowski M, Poordad F, Manns MS, Bronowicki JP, Reddy KR, Harrison SA, et al. Anemia during treatment with peginterferon alfa-2b/ribavirin with or without boceprevir is associated with higher SVR rates: analysis of previously untreated and previous treatment-failure patients. J Hepatol. 2011;54 Suppl 1:S195–6.
17. Telaprevir EU Summary of Product Characteristics.
18. Boceprevir EU Summary of Product Characteristics.
19. Garg V, van Heeswijk R, Lee JE, Alves K, Nadkarni P, Luo X, et al. The effect of telaprevir on the pharmacokinetics of cyclosporine and tacrolimus. Hepatology. 2011;54:20–7.
El tratamiento antiviral para la hepatitis crónica C (genotipo 1) con interferón pegilado (nombres comerciales: Pegasys® y Pegintron®), ribavirina (nombres comerciales Copegus® y Rebetol®) y un inhibidor de las proteasas, boceprevir (nombre comercial Victrelis®) o telaprevir (nombre comercial Incivo®), es efectivo para la curación de la enfermedad en una proporción entre 50 y 75% de los casos, según las características del enfermo (primer tratamiento o fracaso de un tratamiento previo con la combinación de interferón y ribavirina).
Aun así, aunque se respeten sus contraindicaciones, el tratamiento antiviral no está exento de riesgos. Puede causar efectos adversos, de intensidad variable, dependiendo de cada persona, habitualmente benignos y reversibles al dejar el tratamiento sin dejar secuelas, y solo excepcionalmente son graves.
El tratamiento exige visitas médicas periódicas y exámenes analíticos repetidos, para comprobar, en primer lugar, si el tratamiento es beneficioso, y también para detectar, y tratar si es posible, los efectos adversos que se puedan presentar como consecuencia de la medicación, así como evaluar si es conveniente la retirada del tratamiento por carencia de eficacia o por efectos adversos no controlables.
Efectos adversos y/o complicaciones del tratamiento. La mayoría son leves, aunque incómodos, como fatiga, anorexia, dolor muscular, aumento de la caída de cabello, insomnio, dolor de cabeza, irritabilidad, ansiedad, disgeusia (pérdida del sabor), disfunción tiroidea, reducción del número de plaquetas y anemia. La mayoría de estos efectos indeseables se pueden controlar o mejorar con tratamientos sintomáticos. Los efectos adversos más graves son afortunadamente infrecuentes (riesgo aproximado de 1 caso por cada 1.000 enfermos tratados), pero justifican la interrupción del tratamiento y exigen un tratamiento específico. Entre ellos se incluyen: depresión grave, generalmente solo en personas con historia de depresión previa grave; desencadenamiento de una diabetes mellitus; insuficiencia respiratoria; disminución de la agudeza visual o visión borrosa; desencadenamiento de una enfermedad autoinmune o empeoramiento de un psoriasis, y enfermedad cutánea por hipersensibilidad.
Advertencia importante respecto al tratamiento. Este tratamiento puede producir, en caso de embarazo, lesiones o la muerte del embrión o el feto, tanto si es el hombre como si es la mujer quien está tomando el tratamiento. Por lo tanto, el tratamiento antiviral exige que tanto las mujeres en edad fértil como los hombres que conviven con ellas utilicen métodos anticonceptivos eficaces (preferiblemente 2 métodos combinados, por ejemplo preservativo el varón y anovulatorios la mujer) para evitar el embarazo durante todo el tratamiento y hasta 6 meses después de haberlo finalizado. El tratamiento puede provocar también disminución de los niveles de leucocitos o de las plaquetas que pueden obligar a modificar la pauta del tratamiento o a añadir tratamientos adyuvantes para evitar complicaciones.
Alternativas al tratamiento antiviral. Actualmente no hay alternativas de tratamiento para esta enfermedad, y los enfermos no tratados tienen el riesgo de que su enfermedad hepática evolucione a formas clínicas más graves y que aparezcan complicaciones. Si le proponemos hacer el tratamiento es porque hemos considerado que los riesgos y sus inconvenientes son menores que los que puede causar la evolución espontánea de la enfermedad. Sin embargo, la decisión final de seguir o no este tratamiento siempre tiene que ser tomada por el paciente, después de una información precisa de los inconvenientes de la enfermedad y de la medicación que le hará su médico.
Sr/Sra. DNI:
Manifiesta voluntariamente que: He sido informado/informada que el médico que me atiende de la conveniencia de recibir tratamiento antiviral por la hepatitis C que sufro. He comprendido la información que se me ha dado por lo que acepto libremente recibir este tratamiento, sabiendo que me podré desdecir en cualquier momento si así lo decido antes de empezarlo o durante el tratamiento sin necesidad de justificar la razón. Barcelona, dede

