




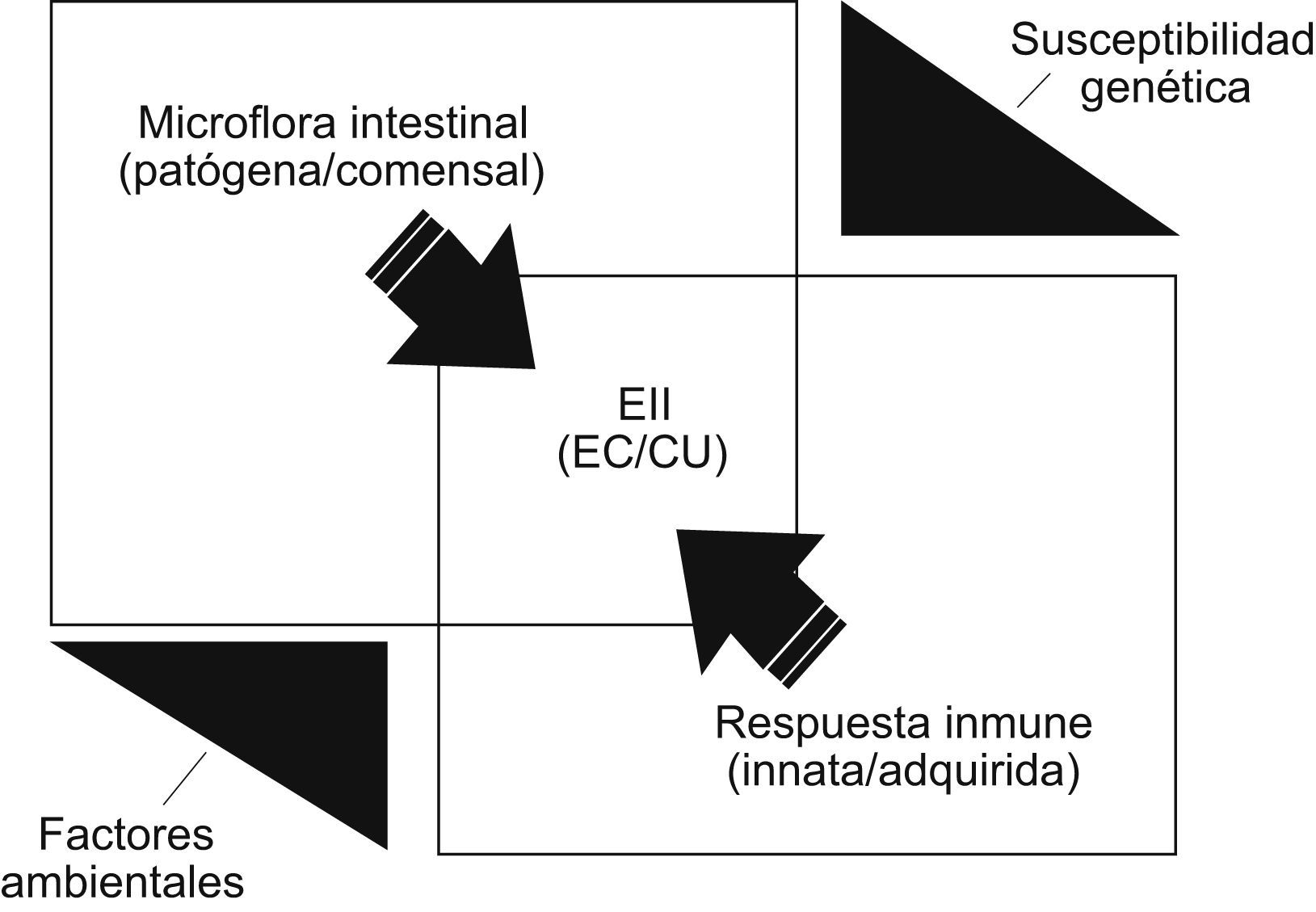
Actualmente todo inclina a pensar que la enfermedad inflamatoria intestinal (EII) en sus 2 variantes, la enfermedad de Crohn (EC) y la colitis ulcerosa (CU), traduce un conflicto entre el sistema inmunitario de la mucosa intestinal y los antígenos intraluminales, fundamentalmente de la microflora intestinal, a los que normalmente toleraba. Todo eso modulado por numerosos factores ambientales y una evidente predisposición de carácter poligénico.
Sobre este argumento se revisa el comportamiento del conjunto de circunstancias etiológicas (microbianas, genéticas y ambientales) para analizar, a continuación, las posibles parcelas patogénicas en donde se expresan aquellos factores etiológicos, como la disfunción del epitelio intestinal, las alteraciones del sistema inmunitario innato y la distorsión de los brazos celular y humoral del sistema inmunitario adquirido. Se comenta brevemente el papel de la isquemia tisular en la EC y la expresión de las “metástasis inflamatorias extraintestinales”, tanto en la EC como en la CU.
Finalmente, se especula sobre la probable consideración de la EII como un espectro de procesos patológicos provocados desde ángulos etiopatogénicos diferentes y el posible significado biológico de su creciente incidencia en el mundo occidental, en coincidencia con el declive de las enfermedades infecciosas en éste.
All the currently available evidence suggests that the two types of inflammatory bowel disease (IBD), Crohn's disease (CD) and ulcerative colitis (UC), involve a conflict between the immune system of the intestinal mucosa and intraluminal antigens, mainly the intestinal microflora, which are normally tolerated by the immune system. This conflict is modulated by numerous environmental factors and a clear polygenetic predisposition. The present article reviews the behavior of all the etiologic circumstances (microbial, genetic and environmental) and subsequently analyzes the possible pathogenic factors in which the etiologies can be found, namely: dysfunction of the intestinal epithelium, innate immune system alterations, and distortion of the cellular and humoral arms of the acquired immune system. The role of tissue ischemia in CD and expression of “extraintestinal inflammatory metastases”, both in CD and UC, are briefly discussed. Finally, the view that IBD may be a spectrum of pathological processes provoked by distinct etiopathogenic factors and the possible biological significance of the growing incidence of this disease in the western world, coinciding with the decline in infectious diseases in this geographical area, are discussed.
Con la denominación un tanto imprecisa de “enfermedad inflamatoria intestinal” (EII) se designa, en patología digestiva, al menos a 2 entidades anatomoclínicas bastante diferentes: la colitis ulcerosa (CU) y la enfermedad de Crohn (EC), a las que algunos autores añaden una tercera variante híbrida —la colitis indeterminada—, que comparte rasgos biológicos de aquellas 21–3. En España, la prevalencia de la EII se mueve entre 200 y 250 casos por 100.000 habitantes; la CU es 2 veces más frecuente que la EC.
La aproximación multidisciplinaria (epidemiológica, histopatológica, genética e inmunológica) a sus 2 fenotipos clínicos principales, junto con los hallazgos obtenidos en diferentes modelos de enterocolitis experimentales (espontáneas, inducidas por agentes químicos, por defectos en la barrera epitelial o por manipulación genética del sistema inmunitario) permiten tener hoy algunas ideas sobre “el cómo” y “el porqué” de éstas4–12. Así, todo inclina a pensar que la EII traduce un conflicto inmunológico entre antígenos (Ag) intraluminales procedentes fundamentalmente de la flora microbiana intestinal y el sistema inmunitario local —tanto innato como adquirido—, el que, por diversas razones, ha perdido su habitual “actitud tolerante” frente a aquel ecosistema microbiano; todo esto bajo una compleja regularización poligénica, con acontecimientos mutacionales que pueden manifestarse en diferentes parcelas patogénicas, modulada por factores ambientales todavía mal conocidos (fig. 1).
 como un conflicto inmunológico entre la microflora intestinal y el sistema inmunitario local al amparo de una susceptibilidad poligénica y modulado por factores ambientales. EC: enfermedad de Crohn; CU: colitis ulcerosa.")
Como consecuencia de aquel conflicto, se pone en marcha una respuesta inmunitaria agresiva y quizá aberrante, que origina una inflamación local crónica y recurrente, con 2 expresiones anatomoclínicas fundamentales. Por una parte, la CU como una inflamación superficial, difusa, continua, simétrica y ulcerohemorrágica a lo largo del tramo colorrectal del intestino. Y por otra, la EC como una inflamación segmentaria, asimétrica, transmural, obstructiva o fistulizante, localizada en cualquier zona del tubo digestivo, con preferencia por el íleon terminal, el colon derecho y la región perianal. Con frecuencia, ambos procesos tienden a expresarse con focos inflamatorios en territorios extraintestinales, lo que muestra una cierta vocación de enfermedad sistémica.
Propósito y estructuración del trabajoDesde una incidencia etiológica multifactorial y una patogenia en la que sus acontecimientos posibles pueden expresarse en diversas parcelas de una secuencia teóricamente multifásica (fig. 2), se va a hacer un desarrollo lo más integrado posible del argumento de este trabajo a en 5 capítulos.

En el primer capítulo se comentará la participación de las circunstancias etiológicas principales (microflora intestinal, predisposición genética y factores ambientales). En el segundo capítulo se analizará el posible papel patogénico que podrían desempeñar ciertos cambios funcionales de la barrera epitelial que separa la luz intestinal de su mucosa. En el tercer capítulo se resumirán los datos que apoyen la participación, en la patogenia de la EII, de las alteraciones de varias parcelas operativas, como el sistema inmunitario innato y los brazos celular y humoral del sistema inmunitario adquirido. En el cuarto capítulo se tratará de ordenar el conjunto de moléculas solubles bioactivas que riegan la pared intestinal en estos procesos y sus efectos mejor conocidos. En un quinto y último capítulo se abordarán algunas parcelas patogénicas marginales, como son los posibles fenómenos isquémicos en la EC y la “metástasis inflamatoria” extraintestinal común a la CU y EC.
A título de epílogo, se intentará recapitular lo que hoy parecen hechos probables así como especular sobre lo que para mañana podrían ser hechos posibles; todo esto, a sabiendas de que ese mañana está por llegar y puede deparar hallazgos sorprendentes que “enreden todavía más la madeja” etiopatogénica y conceptual de la EII, si es que eso es posible.
Circunstancias etiológicasLas circunstancias que ponen en marcha y cronifican la EII en sus 2 formas principales (la CU y la EC) circulan a lo largo de 3 vectores de acción: la microflora intestinal, una cierta predisposición genética y algunos factores ambientales4,6,12. La acción convergente de todos ellos induce una reacción inmunológica enérgica y persistente, cuya consecuencia final es la inflamación crónica de algún segmento del tubo digestivo. A aquellos determinantes etiológicos se dedicarán los apartados siguientes.
Microflora intestinalÉste parece ser —con pocas dudas al respecto— el vector etiológico más importante en la realización de la EII. A continuación se revisará su realidad fisiológica y su posible participación patogénica.
Composición y localizaciónEl intestino humano es el lecho ecológico de una compleja población microbiana que, gracias a las técnicas de identificación molecular, se calcula que supera los 100 billones (1014) de microorganismos, repartidos entre 15.000 y 30.000 especies pertenecientes a más de 1.500 géneros13–15. Esta microflora o microbiota está formada fundamentalmente por bacterias, aunque también la integran virus, hongos y archeas.
La concentración microbiana del contenido intraluminal va aumentando, en cantidad y complejidad, a lo largo del tubo digestivo, al mismo tiempo que se va cambiando de microorganismos aerobios en el estómago y el intestino delgado a bacterias anaerobias en el intestino grueso.
Así, la microflora gastrointestinal se mueve conforme al siguiente gradiente cuantitativo (por gramo de contenido intraluminal) y cualitativo: a) en el estómago (entre 0 y 10 gérmenes/g) está formada por Lactobacillus, Candida, Streptococcus, Helicobacter pylori, Peptostreptococcus, etc.); b) en el duodeno, el yeyuno y el íleon (102gérmenes/g) está integrada por Streptococcus y Lactobacillus; c) en el íleon distal (entre 107 y 108gérmenes/g) predominan Streptococcus, Clostridium, Bacterioides, Corinebacteria, etc., y d) en el ciego y el tracto colorrectal (entre 1011 y 1012gérmenes/g) está integrada por Bacterioides, Clostridium, Bifidobacterium, enterobacteriáceas, etc.
Funciones fisiológicasLa microflora intestinal ejerce, en condiciones normales, importantes funciones13, entre las que destacan: a) degradación de los principios inmediatos que no se han digerido previamente, en la que se obtenienen ácidos grasos de cadena corta y aminoácidos, se producen vitaminas (B12, K, ácido fólico) y se aprovechan elementos minerales (hierro, magnesio, calcio, etc); b) ejerce funciones tróficas sobre la mucosa digestiva, que activan la proliferación y la diferenciación de las células epiteliales; c) favorece el desarrollo y maduración del sistema inmunitario local; d) ejerce un”efecto barrera” al ocupar el lecho ecológico apto para anidar bacterias y evita la implantación de gérmenes patógenos en éste, y, por último, e) secreción de bactericidas por parte de algunos microorganismos residentes amigos, que evita el sobrecrecimiento de bacterias oportunistas al acecho.
Datos que apoyan el papel etiológicoHay toda una serie de datos clínicos y experimentales que indican vehementemente la importancia de la microflora intestinal en la realización de la EII15–23. Entre éstos destacan los siguientes: a) la localización preferente de las lesiones en el íleon terminal y en el colon, donde la densidad microbiana es máxima; b) el parecido histológico de las lesiones granulomatosas de la EC y las de algunas infecciones por micobacterias, entre ellas, la enfermedad de Johne de los rumiantes; c) el que los antibióticos de amplio espectro mejoren la inflamación de la EC ileocólica; d) la posibilidad de disminuir las recidivas postoperatorias de la EC al tratar a los pacientes con metronidazol u ornidazol; e) la prevención de las recidivas de la CU al manipular la flora intestinal con prebióticos; f) el hecho de que, en pacientes con EC, el aislamiento quirúrgico de algún segmento del íleon terminal mejore la lesión inflamatoria, mientras que la reinfusión del contenido intestinal en la zona excluida reactive la inflamación en pocos días; g) el que los pacientes de EII exhiban respuestas serológicas y de sus células T frente a numerosas bacterias entéricas propias, cosa que no ocurre en los sujetos normales; h) el descubrimiento —como se verá más adelante— de que ciertas mutaciones de genes que codifican la síntesis de proteínas receptoras de estructuras moleculares bacterianas por parte del sistema inmunitario innato se asocian a una mayor susceptibilidad de tener EC; i) el que los defectos en la función bactericida de la barrera epitelial del intestino de ratones favorezca el desarrollo de enterocolitis experimentales, y, por último, j) el argumento más convincente a favor del papel etiológico de la microflora intestinal procede de la imposibilidad de producir cualquier modelo de enterocolitis experimental cuando los ratones objeto de éste se estabulan en ambientes asépticos, completamente libres de gérmenes, y bastan sólo unos días en un ambiente normalmente séptico para que la inflamación intestinal se desarrolle en estos roedores; este hecho definitivo justifica la afirmación coloquial entre investigadores del temade que “sin bacterias no hay colitis”, aunque en modo alguno se pueda hablar de “colitis bacteriana”.
Posibles gérmenes culpablesAunque ni la EC ni la CU cumplen los postulados de Koch para que se las considere enfermedades infecciosas propiamente dichas, desde su descubrimiento se han postulado numerosos gérmenes como posibles patógenos causantes de éstas. Así, en algún momento puntual se ha sospechado de microorganismos como Escherichia coli, Clostridium, Campylobacter, Pseudomonas, Yersinia enterocolitica, alguna micobacteria y ciertos virus (citomegalovirus, herpes, etc.). Sin embargo, sólo 2 de estos microorganismos han recibido alguna consideración particular: Mycobacterium paratuberculosis y las cepas enteroadherentes e invasivas de E. coli18,24.
- 1)
La lesión granulomatosa que caracteriza a la inflamación de la EC recuerda mucho a la que provoca el bacilo tuberculoso en los seres humanos y, sobre todo, a la inflamación de la enfermedad de Johne en los rumiantes, provocada por M. paratuberculosis. Fue hace 20 años cuando se cultivó este germen a partir de piezas de resección intestinal de algunos pacientes afectados de EC. A partir de entonces, se han publicado numerosos trabajos en los que se han comunicado datos, poco contrastados y a veces contradictorios, sobre la posible asociación de esta micobacteria y la EC25–30, como el hallazgo de este germen en la sangre, del ácido desoxirribonucleico (ADN) de éste en los tejidos de pacientes afectados de EC o la respuesta favorable (en alguna ocasión) a agentes tuberculostáticos, etc. El hecho de que este germen estuviese presente en la leche pasteurizada procedente de vacadas infectadas y en el suelo en el que se mueven y viven estos rumiantes apuntaba a la posibilidad de una infección zoonótica. Sin embargo, y a pesar de todos estos datos, nunca se han obtenido pruebas histoquímicas de la infección intestinal por parte de este microorganismo en pacientes con EC; el ADN de éste no se ha localizado preferentemente en el íleon terminal de estos pacientes, como cabría esperar; una infección por micobacterias se agravaría al tratarla con fármacos inmunosupresores (corticoides, anti-TNF [tumor necrosis factor antibodies ‘anticuerpos contra el factor de necrosis tumoral’, etc.), cosa que no ocurre en la EC y, por último, cuando se realizó en Australia un estudio ciego bien aleatorizado entre pacientes tratados con una combinación de antibióticos eficaz contra M. paratuberculosis y un placebo, se ha visto que, después de un año de tratamiento, no hay diferencias significativas entre ambas ramas del estudio. A la vista de las numerosas publicaciones sobre esta posibilidad etiológica, la idea más razonable es que quizá este germen puede desempeñar algún papel como inductor de la EC —junto con el conjunto de bacterias comensales entéricas— en un subgrupo de pacientes genéticamente susceptibles, sobre todo en aquellos que exhiben un defecto para eliminar gérmenes de crecimiento intracelular. También podría ocurrir que este germen, relativamente frecuente en ciertos ambientes, colonizase selectivamente la mucosa lesionada de la EC. Pero sea como fuere, parece poco probable que se lo pueda responsabilizar de ésta.
- 2)
En los últimos 10 años, se ha identificado una cepa especialmente virulenta de E. coli en más del 20% de las biopsias mucosas de pacientes con EC, con lesiones recurrentes crónicas en la zona del íleon terminal. Se trata de una cepa enteroadherente e invasiva de E. coli, que sólo se encuentra en el 6% de los controles. Debido a su dotación de pelos tipo 1, estos gérmenes se adhieren con facilidad a las células epiteliales del intestino que, a su vez, muestran una hiperexpresión del receptor membranario específico para esta cepa, la CEACAM-6 (carcinoembryonic antigen-related cell adhesion molecule ‘molécula de adherencia 6 del Ag carcinoembrionario’). Esta adherencia facilita la invasión microbiana de los macrófagos (MFG) residentes en la lámina propia de la mucosa intestinal, hecho que induce en estas células la producción de TNF-α y en los linfocitos T (LT) la secreción de interferón gamma (IFN-γ), citocinas prominentes entre las biomoléculas activas en la EC, como luego se verá31–34.
Además, parece que los pacientes con EC son más sensibles a la infección por este tipo de cepa adherente e invasiva de E.coli, ya que estos sujetos exhiben con frecuencia mutaciones del gen NOD2/CARD15 que codifica, como se comentará más adelante, una proteína receptora específica del muramil dipéptido (MDP) de los peptidoglucanos bacterianos. Aquellas mutaciones deterioran la función defensiva de los MFG frente a bacterias intracelulares, lo que puede originar una acumulación microbiana que induce una respuesta inmunológica del sistema mononuclearfagocítico y la formación de lesiones granulomatosas. A su vez, las citocinas IFN-γ y TNF-α son capaces de inducir una expresión máxima del receptor carcinoembrionario CEACAM-6 en los enterocitos, con lo que se facilita (más si cabe) la adherencia de aquella cepa de E. coli y se cierra todo un “bucle” de acontecimientos, en el que la protagonista es esa cepa hipervirulenta de E. coli presente, a través de su ácido desoxirribonucleico (ADN), en los ganulomas microdisecados de pacientes con EC.
Por tanto, parece que estas cepas adherentes e invasivas de E. coli se asocian con cierta frecuencia a la EC ileal, sobre todo en lesiones recurrentes posquirúrgicas (el 36% de los casos frente al 6% en los casos controles), lo que sugiere un posible papel en la iniciación de la inflamación. En cambio, la prevalencia de este germen en la EC colónica alcanza a menos del 4% de los casos, frente a un 0% en las biopsias de CU y menos de un 2% en los controles. De seguir confirmándose estos hallazgos, podría pensarse en un posible papel etiológico de estas cepas adherentes e invasivas de E. coli en la EC ileal de sujetos genéticamente susceptibles.
- 3)
A pesar de los datos que apuntan a la posible participación de algunos patógenos específicos (M. paratuberculosis y E. coli adherente e invasivo) es evidente que no se está ante una enfermedad infecciosa convencional, y lo más probable es que sea la microflora comensal del intestino, en su conjunto, la que un buen día pierde su actividad de “simbiosis” con el huésped que la aloja y pasa a una actividad de “disbiosis”, lo que pone en marcha una reacción inmunológica agresiva que termina inflamando de manera continua a la pared intestinal, en sujetos genéticamente predispuestos, con la ayuda evidente de numerosos factores ambientales.
Es probable que puedan contribuir a esta “disbiosis” algunas alteraciones discretas en el balance de especies microbianas beneficiosas (como Lactobacillus, Bifidobacterium, etc.) frente a especies peligrosas (Bacterioides, Enterococcus, Klebsiellas, etc.). También pueden contribuir a esta “disbiosis” el descenso de la biodiversidad bacteriana o la adquisición de factores de virulencia, como la hiperexpresión de flagelina por parte de ciertas especies de Clostridium, la adquisición de superoxidodismutasa por parte de Enterococcus faecalis y la misma adquisición de propiedades adherentes e invasivas por parte del E. coli, anteriormente comentada13,14,17,18,35–37.
En la actualidad, nadie duda de que la EII, en sus variantes fundamentales (la EC y la CU), goza de un cierto componente hereditario no mendeliano realizado a través de un impacto poligénico complejo, cuyos protagonistas se están empezando a conocer11,38–42. Los datos de los que hoy se dispone sobre este argumento proceden de 2 fuentes principales: por una parte, la epidemiología descriptiva del proceso y, por otra, la identificación de genes de susceptibilidad.
Epidemiología descriptivaEl perfil epidemiológico de la EII apoya la idea de un significativo factor hereditario en ésta.
Así, en principio se pensó que esta enteropatía inflamatoria era más frecuente en la población caucasiana que entre etnias no caucasianas (asiáticas, afroamericanas, etc.). Sin embargo, con el paso del tiempo se ha ido comprendiendo que esta distinta frecuencia depende, en gran parte, de las diferencias en el estilo de vida y en las condiciones ambientales más que en auténticas diferencias genéticas.
No obstante, lo que sí es evidente es que los sujetos de etnia judía muestran una prevalencia de la EII 2 a 4 veces superior a la de cualquier otro grupo caucasiano y esta prevalencia es más acusada entre judios ashkenazitas que entre sefarditas41–43. Este dato epidemiológico, de origen genético, viene avalado (desde la patología animal) por la incidencia preferente de enterocolitis inflamatorias espontáneas similares a la EII humana en ciertas cepas de ratones (C3H/HeJBIR y Samp1/Yit).
Apunta también, en el sentido de una cierta incidencia hereditaria, el hecho de que la EII se asocie a algunos síndromes genéticos. Así, se sabe que un 3% de los sujetos que presentan síndrome de Turner y un 16% de los sujetos que presentan síndrome de Hermansky-Pudlak presentan concomitantemente una EII44,45.
Es notoria la agregación familiar de la EII, ya que en un 10 a un 20% de los pacientes es posible encontrar algún miembro de la familia afectado y, en ocasiones, se ha comprobado concordancia familiar para el tipo EII e incluso para la localización de ésta41,42,46–48. El riesgo de que se dé este proceso entre los hijos cuando ambos padres lo presentan llega a alcanzar al 33% de ellos.
Los estudios en gemelos son los que han proporcionado más información sobre el peso del vector genético en la realización de la EII. La tasa de concordancia es mucho mayor entre gemelos monocigotos que entre gemelos dicigotos, y es más llamativa en la EC que en la CU41,42,49,50. Así, se ha comunicado una concordancia genética que se sitúa entre el 20 y el 50% para el caso de gemelos monocigotos, cuando uno de ellos desarrolla la EC, y del 10% para el caso de gemelos dicigotos, mientras que para pacientes con CU esta concordancia baja a un 16% para gemelos monocigotos y a un 4% para gemelos dicigotos.
En resumen, se puede decir que el impacto genético es sensiblemente mayor en la EC que en la CU, aunque en gemelos univetelinos raras veces alcanza al 50% de los casos, lo que apunta a la gran importancia de los factores ambientales en la realización de la EII, al margen de la predisposición genética.
Ambas formas anatomoclínicas de EII —la CU y la EC— pueden coexistir en algunas familias. Así, se calcula que el riesgo de que un paciente con CU tenga familiares afectados de EC es del orden de 1,72 veces lo normal, y el riesgo de que pacientes con EC tengan familiares con CU es del orden de 3,85 veces lo normal47. Este hallazgo habla a favor de que ambos fenotipos de EII, a pesar de sus diferencias clinicopatológicas, deben compartir algunos genes de susceptibilidad.
Tal como se verá más adelante, en los pacientes afectados de EII se ha descubierto una serie de marcadores biológicos subclínicos que, de alguna manera, son más o menos específicos de cada fenotipo. Así, se han descrito marcadores de permeabilidad intestinal aumentada en la EC y marcadores séricos en forma de ANCA (antineutrophil cytoplasmic antibodies ‘anticuerpos citoplásmicos antineutrófilos’) con patrón perinuclear (pANCA) en la CU. Todos ellos son más frecuentes en familiares sanos de pacientes con EII que en el resto de la población, incluidos entre esta última los cónyuges de aquéllos, dato que habla de cierta concordancia genética familiar41,51,52.
Identificación de genes de susceptibilidadDurante los últimos 15 años, varios grupos de investigadores han practicado amplios estudios del genoma (“escaneo”) en algunas cohortes de hermanos afectados de determinada forma de EII. De esta manera, se han ido identificando algunas regiones cromosómicas compartidas, en las que se sospecha que puedan estar localizados genes cuya mutación propiciaría el desarrollo del proceso. Estas regiones se han denominado con las siglas IBD (inflammatory bowel disease) y un sufijo numérico correlativo, de acuerdo con el orden cronológico de descubrimiento41.
De esta manera, se han localizado las regiones IBD1 (cromosoma 16q); IBD2 (cromosoma 12q); IBD3 (cromosoma 6p); IBD4 (cromosoma 14q); IBD5 (cromosoma 5q); IBD6 (cromosoma 19p) e IBD7 (cromosoma 1p). De todas maneras, aunque sean importantes estos hallazgos de regiones cromosómicas “sospechosas”, hay que reconocer que se encuentran a gran distancia del objetivo final, que no es otro que la identificación de genes concretos cuya mutación pueda crear un grado mayor o menor de susceptibilidad de desarrollo de algún tipo de EII. La dificultad estriba en el hecho de que en cada una de estas regiones IBD pueden situarse centenares de genes candidatos a desarrollar aquella función patogénica.
Dos fueron los sistemas iniciales de investigación utilizados para la identificación de genes concretos en enfermedades de etiología multigénica: la clonación posicional, mediante la utilización de estudios de “ligamiento”, y la aproximación a genes presuntamente candidatos, mediante la utilización de estudios de asociación de casos y controles. Con estas herramientas de trabajo se empezó a localizar algunos genes que parecen estar implicados en la inducción de alguna forma concreta de EII, a veces limitada a un colectivo étnico singular, como se verá a continuación.
En la región IBD1 se ha identificado el primer gen relacionado con la susceptibilidad de presentar EC. Se trata del llamado gen NOD2 (nucleotide oligomerization domain 2) actualmente rebautizado como CARD 15 (caspase recruitment domain 15), localizado en el cromosoma 16q1.2. En condiciones normales, el gen NOD2/CARD15 codifica la síntesis de una proteína que actúa como receptora citosólica, en células del sistema inmunitario innato (MFG y células dendríticas [CD]) de peptidoglucanos bacterianos a través de su componente MDP.
En el año 2001, 2 grupos de investigadores comunicaron la asociación entre mutación(es) de ese gen y la aparición de una EC de localización ileal y evolución fibroestenótica22,23. Aunque se ha descubierto una treintena de posibles mutaciones del gen NOD2/CARD15, hoy se sabe que sólo 3 de ellas se asocian a entre un 25 y un 35% de pacientes de ancestro europeo afectados de EC, sin que esta asociación se dé en pacientes de etnias asiáticas ni tampoco en sujetos afectados de CU41,53–58. Estas mutaciones se producen por la sustitución de un solo aminoácido en una región rica en repeticiones de leucina responsable del reconocimiento de la molécula MDP, que es la “zona biológica activa” de los peptidoglucanos bacterianos.
Parece que cuando se presenta una mutación de este gen, el riesgo de tener una EC es de 2 a 3 veces lo normal; pero este riesgo sube de 20 a 40 veces lo normal cuando se presentan 2 mutaciones. Al ser la prevalencia de la EC en los países de Europa occidental del orden de uno a 2 casos por cada 1.000 habitantes, se puede calcular que la susceptibilidad de presentar una EC con 2 mutaciones del gen NOD2/CARD15 oscila entre un 4 y un 8% de los sujetos. A la vista de estos cálculos de epidemiología genética, puede afirmarse que se está ante un acontecimiento mutacional con poca penetrancia, dado que en menos del 10% de las personas portadoras de 2 mutaciones del citado gen se desarrolla una EC. Este hecho apunta con claridad a que son necesarias otras anomalías genéticas de susceptibilidad y, sobre todo, del efecto de factores ambientales, para poner en marcha el proceso inflamatorio de la EC.
Tres años después, en el año 2004, se descubrió que 2 variantes funcionales de los genes OCTN (organic cation transporter)1 y OCTN2, localizados en la región IBD5 (cromosoma 5q3.1), se asocian a una susceptibilidad incrementada de tener una EC por parte de los individuos que presentan acontecimientos mutacionales en el segmento de transcripción del primero y en la región promotora del segundo. Ambos acontecimientos afectan a la transcripción y función de las proteínas transportadoras de cationes orgánicos59. En el mismo año 2004, se identificó un tercer gen también relacionado con la EC. Se trata del gen DLG5, localizado en el cromosoma 10, que en condiciones normales codifica la síntesis de una proteína que asegura el andamiaje celular y que parece ayudar a mantener la integridad epitelial. Se han encontrado variaciones mutacionales de este gen en el 10% de los pacientes afectados de EC60.
Algunas variantes del gen PPARG (peroxisome proliferative-activated receptor gamma) parecen ser las causantes de la colitis crónica esporádica que tienen los ratones de la raza SAMP1/Yit. Igualmente, parece que algunos raros polimorfismos de este gen, situado en los seres humanos en el cromosoma 3, se asocian de alguna manera a la EC humana11,60. Es bien conocido que la proteína codificada por el gen MDR1 (multidrug resistance gene 1) actúa normalmente como un transportador que rige el flujo transmembranario de fármacos en las células. Algunas variantes mutacionales de este gen parecen comportarse como un factor de susceptibilidad de la CU11,60.
En la región IBD3 (cromosoma 6p1.3) están situados los genes que codifican la síntesis de los HLA (human leukocyte antigen ‘antígenos de histocompatibilidad’) de clase II y los de la superfamilia TNF. Al revés de lo que ocurre con otras enfermedades de patogenia inmunitaria (artritis reumatoide, diabetes mellitus tipo 1, esclerosis múltiple, etc.), la asociación entre genes del sistema HLA y la incidencia de la EII es poco consistente. De todas maneras, la CU se ha asociado al genotipo HLA-DR2 (DRB1* 1502) en pacientes japoneses, mientras que en enfermos europeos la CU parece asociarse discretamente al genotipo HLA-DR3 (DRB1* 0103). Las asociaciones de los Ag de clase II del sistema HLA a la EC son menos convincentes, aunque parece que el genotipo HLA-DR1 se ha relacionado con esta variante de la EII4,41,60.
Finalmente, el gen NOD1/CARD4, localizado en el cromosoma 7p1.4, codifica la síntesis de una proteína que se comporta normalmente como un receptor citosólico con función parecida a la del receptor NOD2. Parece que un polimorfismo complejo de este gen se asocia a un incremento de susceptibilidad de presentar tanto EC como CU60.
En los últimos 3 o 4 años, el conocimiento creciente del genoma humano (Human Genome Project), junto con la puesta a punto de una tecnología de genotipaje de “alto rendimiento”, está permitiendo realizar investigaciones genéticas en gran escala. De esta manera, se están descubriendo con gran rapidez qué polimorfismos o variantes genéticas se asocian a un cierto riesgo de presentar enfermedades de etiología compleja (poligénica y ambiental), como el asma, la diabetes, el cáncer, la cardiopatía isquémica y, por supuesto, la EII. Se está haciendo referencia a los programas de investigación conocidos como Genome-wide Association Studies (identificados con el acrónimo GWAS), que posiblemente conducirán a dibujar la “arquitectura genética” de procesos tan complejos como la EII61.
Con la tecnología GWAS, aparte de confirmar el papel de susceptibilidad de algunos polimorfismos genéticos identificados en la era previa (genes NOD2/CARD15, DLG5, etc.), se han descubierto otras nuevas asociaciones60,62–66.
De esta manera, se conocen ya, en el momento actual, cerca de 40 genes y sus polimorfismos o variantes incrementan el riesgo de desarrollo de una EC en las personas que los exhiben en su mapa genético. Entre estos nuevos genes descubiertos con la tecnología GWAS e implicados en la EC se encuentran el gen TNF-SF15 (cromosoma 9q3), perteneciente a la superfamilia de genes TNF; el gen IL-23R (cromosoma 1p3) que codifica la síntesis del receptor de la citocina IL-23 (su importante papel en la diferenciación de la línea celular Th-17 se comentará más adelante); los genes implicados en la autofagia, ATG16L1 (cromosoma 10q2) e IRGM (cromosoma 5q3), cuyas proteínas normales desempeñan un papel importante en la defensa antimicrobiana del sistema inmunitario innato, inducen la autofagia celular y, con esto, la eliminación de patógenos intracelulares (micobacterias, etc.), acontecimiento que no realizan algunos de sus polimorfismos; también parecen desempeñar algún papel los genes PTGR4 (cromosoma 5p1), MST1 (cromosoma 3p2), PTPN2 (cromosoma 18p1), etc., y así hasta más de 30 genes que pronto serán muchos más.
Algo menos de un tercio de estos genes cuyos polimorfismos se asocian a la EC parece que predisponen también a presentar CU. Entre ellos destacan el citado gen IL-23R, comentado anteriormente, y otros menos conocidos, como los genes IL-12B (cromosoma 5q3), JAK2 (cromosoma 9p2), STAT-3 (cromosoma 17q2), etc.
Con la tecnología GWAS se están empezando a localizar algunos polimorfismos que podrían actuar como inductores selectivos de la CU en el cromosoma 1p36 (en los genes OTUD3 y PLA2G2E) y del cromosoma 12q15 (genes de las IL-22, IL-26 y del IFN-γ).
El tremendo giro tecnológico que está significando la “era de los GWAS” para la búsqueda en gran escala de polimorfismos genéticos implicados en la susceptibilidad de presentar la EII está empezando a dar sus primeros rendimientos, como confirmar el carácter altamente poligénico de esa susceptibilidad; confirmar, igualmente, que junto con genes que parecen participar en la realización exclusiva de la EC o de la CU, hay otros que participan de manera inespecífica en la EII en su conjunto; subrayar que la carga genética de la EC es muy superior a la de la CU; descubrir que, a pesar de la escasa penetrancia de los polimorfismos del gen NOD2/CARD15 en la inducción de la EC, ellos solos son los causantes de más del 20% de la predisposición genética de ésta. De todas maneras, habrá que esperar unos pocos años para valorar el alcance de lo que se va a conocer sobre cómo se realiza ese amplio espectro de procesos en los que se está convirtiendo la llamada EII.
Factores ambientalesEl tercer vector por el que circulan otras circunstancias etiológicas que influyen en el desarrollo de la EII está formado por el amplio manojo de factores ambientales o factores de riesgo externo que modulan la respuesta inflamatoria, al alza o a la baja42,67–69. La importancia de este vector etiológico viene avalada por 3 hechos. En primer lugar porque, aunque es notable la predisposición genética en la realización de la EII —sobre todo en la EC—, raras veces, en este último proceso, se alcanza el 50% de concordancia entre gemelos monocigotos. En segundo lugar, por las modificaciones que las emigraciones de poblaciones humanas irroga sobre la prevalencia de la EII en éstas, prevalencia que se aproxima a la del país de acogida. Y en tercer lugar, por el progresivo incremento de la incidencia de esta enfermedad en los países en desarrollo, a medida que su estilo de vida se ha ido “occidentalizando”.
Hechas estas consideraciones, se intentará resumir lo que se conoce —que no es mucho— y lo que se sospecha —que es bastante— sobre este argumento.
- 1)
La lactancia materna estimula la maduración y el desarrollo de la mucosa del tubo digestivo, y algunos estudios indican que el haber recibido esta alimentación posnatal aminora el riesgo de tener EII70. Sin embargo, los resultados de este estudio no se han confirmado.
- 2)
Hace años, algunos autores pensaron en la posibilidad de que en la patogenia de la EC desempeñase un papel un factor isquémico provocado por infartos intestinales multifocales. Al buscar motivación sobre este acontecimiento, se sugirió la posibilidad de que estas lesiones isquémicas fuesen secundarias a una vasculitis granulomatosa producida por una infección pesistente por el virus del sarampión71–74. Sin embargo, las pruebas epidemiológicas, inmunohistoquímicas o serológicas a favor de este argumento han sido muy dispares y, en general, poco convincentes. Por otra parte, parece que el antecedente de gastroenteritis infecciosa aguda incrementa el riesgo de tener una EII75.
- 3)
Por el contrario, se ha especulado con la posibilidad de que algunos factores ambientales podrían actuar como “protectores” contra el desarrollo de la EII. Así, se ha sugerido que el contacto con animales estabulados en granjas (ganado vacuno, caballar, etc.) durante los primeros años de la vida, disminuye el riesgo de tener una EII68,69. Esta observación no confirmada, casa con la hipótesis de que la exposición a parásitos (helmintos, etc.) y microorganismos potencialmente patógenos de los que son portadores aquellos animales, en la primera infancia, confiere protección a estos niños de tener enfermedades mediadas por reacciones inmunitarias (alérgicas, inflamatorias o autoinmunitarias), tal como sugiere la llamada “hipótesis de la higiene”, de la que se hablará al final del trabajo.
- 4)
En un mataanálisis publicado en el año 2000 que recoge los resultados de 17 estudios epidemiológicos de tipo casos y controles, se ha concluido que la apendicectomía practicada en la infancia disminuye en un 69% el riesgo de desarrollo posterior de una CU, pero es muy dudoso el efecto de esta intervención sobre la incidencia de una EC42,76–78. Lo mismo ocurre en el modelo experimental que más se parece a la CU humana: los ratones deficientes en el TCR-α (T cell receptor alpha ‘receptor alfa de las células T’), en los que se desarrolla una colitis espontánea a las 3 o 4 semanas de vida, acontecimiento que se evita practicando la apendicectomía precoz.
- 5)
El tabaquismo, en forma de hábito de fumar cigarrillos, es el factor exógeno que muestra un efecto más claro sobre la incidencia de la EII, y curiosamente este efecto es opuesto sobre la CU y la EC. En el mejor metaanálisis publicado sobre este tema, que comprende 9 estudios de pacientes con EC y 13 estudios de casos de CU, se ve con claridad que el hábito sostenido de fumar incrementa el riesgo de tener la EC al doble de lo normal y, por el contrario, aminora el de tener la CU a la mitad de lo normal. Cuando se abandona este hábito se invierten aquellas tendencias42,79–81.
- 6)
Se han realizado numerosos estudios epidemiológicos en busca de la posible existencia de alguna relación entre la toma persistente de anticonceptivos orales y el desarrollo de la EII82–84. Se pueden resumir los hallazgos de estos trabajos diciendo que con la primera generación de anticonceptivos, tomados en las dosis habituales de aquella época, parece que había una cierta asociación con el desarrollo de una EC. Por el contrario, este efecto es más dudoso con las dosis menores utilizadas con los anticonceptivos de la segunda y tercera generación.
- 7)
Se ha sospechado el posible papel etiológico del algunos componentes de la dieta42,85–89. Así, se ha comentado que la inclusión en el régimen alimentario de azúcar refinado propicia el desarrollo de la EC, y el uso de margarina en exceso se ha asociado al desarrollo de la CU. Una dieta pobre en verduras y fruta aumentaría el riesgo de EC. Lo mismo ha ocurrido en Japón, donde la incidencia de EC ha aumentado en estos 50 últimos años, lo que ha coincidido con el descenso del consumo de pescado (rico en grasas poliinsaturadas omega-3) y su sustitución por proteínas cárnicas (ricos en ácidos grasos poliinsaturados omega-6). Sin embargo, hoy no se tienen pruebas concluyentes de que la dieta desempeñe algún papel en la etiología de la EII.
- 8)
Es controvertido el posible efecto adverso de la toma crónica de antiinflamatorios no esteroides (AINE) en la incidencia de la EII o en la exacerbación de ésta, efecto sorprendente para unos fármacos a los que se califica de “antiinflamatorios”90,91. Es posible que este hecho guarde relación con el aumento de la permeabilidad del epitelio intestinal que provocan estos fármacos.
- 9)
Cada vez se conoce mejor que las entidades que integran la EII son más prevalentes en países desarrollados del llamado mundo occidental que en países subdesarrollados. Este hecho podría guardar relación con el mejor nivel higiénico y sanitario de los primeros y, con esto, con un descenso drástico de infecciones parasitarias (helmintos y protozoos) que suelen originar respuestas inmunitarias de tipo Th2, con secreción de citocinas antiinflamatorias (IL-4, IL-5 e IL-13). En ausencia de este tipo de acontecimiento inmunológico sería más fácil el desarrollo de una EII (“teoría de la higiene”)92,93.
El despliegue virtual de la barrera que forma el epitelio intestinal en un ser humano adulto representa una superficie aproximada de 300 a 400m2. En condiciones funcionales normales, esta extensa frontera representa una “interfase inteligente” entre 2 mundos condenados a vivir en paz: el de los Ag microbianos y alimentarios, por parte del espacio intraluminal, y el del sistema celular inmunocompetente (innato y adquirido), por parte del espacio mucoso.
Cuando por alguna razón se deteriora esta barrera, aquellos Ag atraviesan el epitelio intestinal “a destajo” y se rompe el estado de “tolerancia inmunitaria”, que permitía la coexistencia pacífica de aquellos 2 mundos. Al ocurrir esto, se provocan respuestas inmunitarias capaces de originar la inflamación crónica de la pared intestinal que caracteriza al proceso que es objeto del presente estudio93,94. Se recordará en los apartados de este capítulo, la histología funcional de la barrera intestinal y se comentarán brevemente los datos clínicos y experimentales que apoyan la sospecha de que algún tipo de distorsión estructural o funcional de ésta podría convertir a esta barrera en una “parcela patogénica” que explique la realización de la EII o de alguna de sus variantes en determinadas circunstancias genéticas o ambientales.
Histología funcional del epitelioEl epitelio intestinal está formado por una monocapa de células conectadas entre sí por una red de moléculas de adherencia que sellan los espacios intercelulares, lo que contribuye a hacer muy selectivo el paso transepitelial de moléculas y microorganismos desde la luz intestinal. La mayoría de las células que integran este epitelio están especializadas en la absorción de nutrientes y, salpicando su abigarrado despliegue, se sitúa una segunda familia de células caliciformes secretoras del moco que actúa como lubricante de la superficie epitelial. En zonas muy concretas, como es la cúpula que recubre los amasijos linfoides que forman las placas de Peyer, se sitúa un tercer tipo de células epiteliales, las llamadas células M, que cumplen el papel de introductoras de Ag intraluminales hacia el MALT (mucosa-associated lymphoid tissue ‘tejido linfoide asociado a la mucosa’). Por último, en el fondo de las criptas del intestino delgado se encuentran situadas la células de Paneth, pertenecientes a una cuarta familia de elementos celulares, y su misión principal es segregar péptidos bactericidas (defensinas y criptidinas) que, junto con la inmunoglobulina (Ig) A dimérica segregada por los plasmocitos subepiteliales, contribuyen a neutralizar a cualquier microorganismo intraluminal que ponga en peligro la “simbiosis” de la microflora residente en el intestino con el huésped que la alberga1,39,93,95.
Datos aportados por la patología humanaEl estudio de la EII ha aportado algunos hallazgos que apoyan el papel patogénico de la disfunción epitelial en su gestación.
- 1)
Así, tanto en pacientes afectados de EC como en sus familiares directos asintomáticos, se ha detectado un incremento de la permeabilidad intestinal, lo que indica una disfunción epitelial de naturaleza genética 11,39,94,96,97. Esta disfunción no se ha detectado en la CU.
Nadie duda de que la inflamación de la mucosa del intestino delgado, sea cual fuere su origen, podría irrogar como epifenómeno una lesión de su epitelio con aumento de su permeabilidad. Sin embargo, lo que es más “sugerente” es que este hecho puede ser un acontecimiento primario en el camino patogénico en la EC. No se conoce bien cuál podría ser el mecanismo molecular de esta disfunción epitelial; quizá pueda tratarse de un cambio mutacional de alguno de los genes que codifican la síntesis de los diferentes componentes de la molécula de claudina implicada en las conexiones interepiteliales.
- 2)
En apoyo de lo comentado en el punto anterior se encuentra el conocimiento de que ciertos acontecimientos genéticos concretos incrementan el riesgo de presentar una EC o una CU a través de mutaciones de genes que codifican la síntesis de proteínas implicadas en el mantenimiento de la integridad funcional del epitelio intestinal38,39,59,60,62. Así ocurre con los genes OCTN1 y OCTN2, cuyas proteínas se comportan como eficaces transportadoras de cationes orgánicos a través del epitelio; con el gen DLG5, que codifica la síntesis de la guanilato-cinasa, molécula responsable del mantenimiento de la polaridad de las células epiteliales y con esto de su integridad, y con el gen MDR1, cuya proteína gobierna el flujo transmembranario de fármacos en las células.
El común denominador de las mutaciones de estos genes es que, de alguna manera, facilitan el desarrollo de la EC (genes OCTN y gen DLG5) o de la CU (gen MDR1) a través de un cierto grado de disfunción epitelial.
- 3)
Mención especial merece el gen NOD2/CARD15 que, como se vio en el capítulo anterior, codifica la síntesis de una proteína receptora de los peptidoglucanos bacterianos a través de su componente MDP, y que mutaciones de este gen propician la aparición de la EC de localización ileal en colectivos de pacientes caucasianos. Pero además, se sabe que la proteína NOD2 se expresa también en las células de Paneth del intestino delgado y que, de alguna manera, su correcta expresión favorece la secreción de defensinas por parte de esta familia de células epiteliales. Ciertas mutaciones del gen NOD2/CARD15 no sólo deterioran el reconocimiento bacteriano por parte de MFG y CD, sino que también disminuye la secreción de defensinas beta-2, beta-3, alfa-5 y alfa-6 por parte de las células de Paneth ileales. Sobre la base de estos hallazgos se ha sugerido la posibilidad teórica de que la EC fuese un “síndrome de inmunodeficiencia especial” por déficit de defensinas11,39,98–102.
- 4)
Parece que el efecto adverso de la administración de AINE (aspirina, indometacina, etc.) a pacientes con EC se podría atribuir al incremento de la permeabilidad intestinal que provocan estos fármacos11,94,103. El hecho de haber encontrado, en alguna ocasión, defectos de esta permeabilidad en parientes asintomáticos de enfermos con EC, tras la administración de AINE, indica la posibilidad de un trastorno genético primario.
- 5)
En algún momento se ha citado la existencia de una capa de moco anormalmente delgada sobre el epitelio colónico de pacientes con CU104. De confirmarse este hecho, podría deteriorar la permeabilidad del epitelio colorrectal en esta EII y quizá se explicaría el efecto beneficioso que sobre ella ejerce el tabaquismo mantenido a través de un incremento de la secreción de ese moco, por las células caliciformes, inducida por la nicotina.
- 6)
La hiperexpesión de la estructura molecular CEACAM-6 (relacionada con el Ag carcinoembrionario) en las células del epitelio ileal de pacientes con EC facilita la ligazón y posterior penetración de cepas de E. coli “adherente e invasivas” en estos pacientes, con probable efecto patógeno sobre la pared intestinal. Es posible que la hiperexpresión de aquella molécula receptora sea inducida directa o indirectamente por la propia cepa de E. coli. Pero, sea como fuere, es un nuevo ejemplo de cómo las modificaciones funcionales de la barrera epitelial pueden influir, en algún momento, en la patogenia de la EII33,34,105,106.
La realización de algunas enterocolitis crónicas más o menos parecidas a la EII humana es también debida a lesiones del epitelio intestinal de los ratones involucrados en alguno de los modelos experimentales8,10.
- 1)
Así, hoy se sabe que la ileítis espontánea que se desarrolla en los ratones de la cepa SAMP-1/Yit, a las 10 semanas de vida, es debida a una deficiencia congénita de la claudina-2, molécula involucrada en la conexión interepitelial de los enterocitos.
- 2)
La ablación del gen que codifica la síntesis de la N-caderina en ratones es la causante de un modelo experimental de colitis espontánea. La ausencia de una molécula tan importante para el ensamblaje celular como la N-caderina origina, sin duda, una disfunción epitelial inductora de la inflamación colónica.
- 3)
Un incremento de la permeabilidad intestinal parece ser el causante de las colitis experimentales en ratones por déficit del gen de la IL-10 y del gen MDR1.
- 4)
Otro ejemplo de colitis experimental murina por lesión tóxica de la barrera epitelial es la que provoca la administración continuada de sulfato sódico de dextrano.
Todos los modelos experimentales comentados en este apartado muestran, de manera clara, el papel que una barrera intestinal deteriorada por distintos caminos puede desempeñar en la realización de algo parecido a lo que, en los seres humanos, es la EII. Parece lógico extrapolar estos hallazgos en apoyo del argumento defendido en este capítulo; es decir, que en la barrera intestinal se puede desarrollar una parcela patogénica importante para algunas variantes etiológicas de la EII humana.
Participación del sistema inmunitarioAl otro lado de la frontera epitelial, en la mucosa intestinal, se sitúa el sistema celular inmunocompetente que hace frente al desafío constante de los Ag intraluminales, fundamentalmente microbianos. Este dispositivo celular se despliega en 2 líneas defensivas que actúan escalonadamente: una primera línea de acción inmediata formada por las células encargadas de la llamada inmunidad innata, pertenecientes de manera dominante al sistema mononuclearfagocítico. Una segunda línea, de acción retardada, es responsable de la llamada inmunidad adquirida o adaptativa y su labor corre a cargo de la población celular linfoide, que trabaja desde sus 2 brazos funcionales responsables de la inmunidad adquirida celular que realizan los LT y de la inmunidad adquirida humoral que protagonizan los linfocitos B (LB). Una cierta proporción de mastocitos y granulocitos eosinófilos completa la discreta expresión de células residentes inmunorreactivas que traducen la situación de “inflamación fisiológica permanente” en la que vive la mucosa intestinal como consecuencia de la vecindad del mundo antigénico intraluminal95,107. En los 3 apartados de este capítulo se hablará del comportamiento de estas áreas de la inmunidad en la EII.
Sistema inmunitario innato y enfermedad inflamatoria intestinalLa llamada inmunidad innata es realizada por parte de un sistema celular inespecífico, independiente del Ag, que actúa como una primera línea defensiva frente a microorganismos intestinales que, por alguna razón, atraviesan la frontera epitelial del tubo digestivo. Se trata de una forma de inmunidad muy primitiva que surgió en la evolución de las especies animales hace millones de años como un primer mecanismo de autodefensa108,109.
Citología funcionalForman parte de este sistema los monocitos MFG y CD, aunque las mismas células epiteliales del intestino, de manera extraoficial, cumplen a veces misiones propias de la inmunidad innata109,110.
- 1)
Estratégicamente, este conjunto de células actúa frente a los microorganismos invasores mediante el desarrollo de una serie de proteínas que se comportan como receptores de determinadas estructuras microbianas conocidas como PAMP (pathogen-associated molecular patterns ‘patrones moleculares asociados a patógenos’), que se comportan como “ligandos específicos” de aquellos receptores de localización transmembranaria y citosólica93,108,109.
Entre los PAMP mejor conocidos se encuentran los peptidoglucanos, que forman parte de la membrana de la mayoría de las bacterias aerobias y anaerobias; los lipopolisacáridos, endotoxinas presentes en las bacterias gramnegativas y por tanto presentes en más del 50% de las bacterias intestinales; las flagelinas, que son un componente de los flagelos bacterianos y un factor de virulencia de bacterias grampositivas y gramnegativas; también pertenecen a estos patrones moleculares bacterianos los lipopéptidos, los ácidos lipoteicoicos, los glucanos y el ADN de doble cadena.
Los receptores de estos PAMP se agrupan en 2 familias de proteínas fundamentales: los llamados TLR (Toll-like receptors ‘receptores tipo Toll’), de localización transmembranaria, y las proteínas NOD, de localización citoplasmática.
- 2)
El primer TLR se identificó en la mosca drosófila, y se comprobó después su presencia en las células del sistema inmunitario innato de los mamíferos; entre ellos, en el hombre, en localización transmembranaria93,95,108,109,111,112. Hasta el momento se ha identificado una docena de TLR en los mamíferos, entre los que destacan, por ser mejor conocidos sus ligandos específicos, los TLR-1 y TLR-2 (peptidoglucanos y lipopéptidos); el TLR-3 (ADN de doble cadena); el TLR-4 (lipopolisacáridos); el TLR-5 (flagelina), etc.
- 3)
Las proteínas NOD constituyen un grupo de receptores de PAMP que, a diferencia del anterior, tiene una localización citosólica en las células del sistema inmunitario innato; hasta la actualidad, se ha descubierto una veintena de éstos. El receptor NOD-2 ha sido, con mucho, el más investigado en relación con la EII de la que se ocupa el presente estudio. Esta proteína NOD-2 actúa como receptora del MDP que, como se vio anteriormente, es una subestructura molecular relacionada con los peptidoglucanos bacterianos93,95,108,109.
- 4)
Por sus ligandos respectivos, la estimulación de los TLR y las NOD tiene 3 consecuencias fundamentales93,108: por una parte, incrementa la capacidad fagocítica de las células portadoras de estos receptores (MFG, etc.); en segundo lugar, pone en marcha las vías intracelulares de “transducción de señales”, tales como las que protagonizan el NF-kB (nuclear factor kappa B ‘factor nuclear kappa B’) y las proteincinasas activadas por mitógenos, que tienen como finalidad activar intranuclearmente determinados genes que codifican la síntesis de citocinas y quimiocinas proinflamatorias; en tercer lugar, aquella estimulación microbiana ayuda a “madurar” a las células presentadoras de Ag (CPAg) con el fín de que éstas transmitan adecuadamente las señales de alarma a las células del sistema inmunitario adquirido, fundamentalmente a los LT.
- 5)
La presencia masiva y continua de Ag intraluminales (flora microbiana comensal, patógenos eventuales y proteínas alimentarias), sólo separados de la lámina propia de la mucosa por la extensa frontera epitelial, requiere una “gestión inteligente” por parte de las CPAg profesionales, como son las CDs, y de sus compañeros de trabajo, los MFG y células epiteliales.
La misión inicial de este colectivo de CPAg es el de “valorar” la calidad de los Ag, que tiene que procesar y ofertar al sistema inmunitario adquirido, con el fín de distinguir los epítopos inofensivos de los epítopos dañinos. Frente a los primeros habrá que mantener una actitud de “tolerancia inmunitaria”, mientras que frente a los segundos será necesario montar una “respuesta inmunitaria” defensiva “antigenoespecífica”, que correrá a cargo de la inmunidad adquirida, tanto celular (LT) como humoral (LB y células plasmáticas). Por esto, de alguna manera, las CD representan un “puente de unión” entre los sistemas inmunitarios innato y adquirido. Los péptidos antigénicos procedentes de la luz intestinal, después de ser debidamente procesados por las CD, se ligan a los Ag de clase II del sistema mayor de histocompatibilidad (DP, DQ y DR), situados en la membrana de aquellas células, y se ofertan fundamentalmente a los LT de fenotipo “colaborador” (LT-CD4+) de su entorno, que los reciben a través de sus receptores membranarios; se inicia, así, una clonogénesis reactiva “antigenoespecífica”.
Una serie de hallazgos realizados estos últimos años están empezando a hacer ver la participación del sistema inmunitario innato en la realización de la EII, con lo que se vislumbra una parcela patogénica que promete ser importante a medio plazo5.
- 1)
Como se vio en un capítulo anterior, se ha comunicado por parte de algunos grupos de trabajo la asociación entre ciertas mutaciones del gen NOD2/CARD15 y la susceptibilidad de presentar EC. Así ha ocurrido entre un 25 y un 35% de los pacientes de ancestro europeo afectados de EC22,23,40,58,113,114. Como se recordaba anteriormente, la proteína NOD2 actúa como receptora intracitoplásmica del MDP, derivado de los peptidoglucanos de la pared de la mayoría de las bacterias, y con esto se activa el NF-κB que induce a una serie de genes para codificar la síntesis de citocinas y quimiocinas proinflamatorias.
No se conoce con seguridad de qué manera la mutación del gen NOD2/CARD15 puede favorecer la realización de la EC, así como ocurre en una pequeña fracción de los pacientes (menos del 10% de los que exhiben 2 mutaciones del gen). Así, mientras los estudios in vitro hablan de que estas mutaciones originarían una “pérdida de función” del receptor y con eso una falta de activación del NF-κB, los estudios in vivo dicen todo lo contrario, es decir, que parece haber una “ganancia de función” de aquel receptor con un incremento en la actividad de aquel factor nuclear, con la consiguiente hiperproducción de citocinas proinflamatorias11,115. Datos proporcionados por algún modelo experimental podría ayudar a dar la solución a este dilema, ya que se ha visto que el TLR2 en ratones dirige a la baja la respuesta del NF-κB. Sin embargo, cuando hay un déficit del gen NOD2 en estos roedores, desaparece esta inhibición sobre el citado factor nuclear, con esta “desinhibición” se producen valores elevados de éste y, con esto, una síntesis exagerada de citocinas inflamatorias patogénicamente agresivas, tal como ocurre en la EC116,117. La extrapolación de este hallazgo a los seres humanos no sólo ayuda a resolver el problema de la interpretación del papel de las mutaciones del gen NOD2/CARD15 en la patogenia de la EC, sino que pone sobre el tapete de la investigación biológica la oscura interrelación entre las proteínas de las familias “de tipo Toll” y NOD, tal como parece también ocurrir normalmente entre TLR9 y NOD2, y que se pierde en la EC asociada a mutaciones del gen NOD2/CARD15118.
- 2)
También se han comunicado alteraciones puntuales en la expresión de algunas proteínas de las familia TRL en la EII11,40,119,120. Así, parece que la proteína TLR-3 se expresa de manera reducida en las células epiteliales intestinales de la EC, sin modificación sensible en la CU. Por otra parte, se ha comunicado una hiperexpresión de la proteína TLR-4 en las células epiteliales del intestino, tanto en la EC como en la CU. Y también se ha publicado que la presencia de ciertos polimorfismos genéticos de los receptores TLR-1, TLR-2, TLR-3, TLR-4, TLR-6 y TLR-9 se asocian a la EII. De todas maneras, hoy se desconoce el papel patogénico que podrían desempeñar todas estas variantes mutacionales en la EII.
- 3)
En el apartado de genética del capítulo de etiología se han resumido la asociación encontrada por algunos autores entre ciertos serotipos y genotipos del sistema HLA y la predisposición a tener EC o CU en determinadas etnias11,40. Hay que confesar de entrada que estos hallazgos no se han confirmado del todo. Sin embargo, no repugna a la lógica el pensar que ciertas modificaciones en la operatividad de los Ag de clase II del sistema HLA situados en la membrana de las CPAg (CD, MFG, etc.) del sistema inmunitario innato podrían “distorsionar” la oferta antigénica al brazo celular de la inmunidad adquirida, con efectos lesivos sobre el entorno celular. Pero esto no está probado.
La segunda línea defensiva del sistema inmunitario intestinal frente a Ag intraluminales (microbianos, etc.) está formada por el conjunto de células que integra el MALT, dividido en los brazos celular (células T) y humoral (células B) del sistema inmunitario adquirido95,121,122. En este apartado se resumirá la citología funcional de la población linfoide encargada de las respuestas inmunitarias de tipo celular y se comentará la participación de esta vertiente del sistema inmunitario adquirido en la EII, conocida a partir de los estudios en patología humana y deducida de algunos modelos de enterocolitis experimentales que recuerdan a la EC o a la CU.
Citología funcionalLas respuestas inmunitarias de carácter celular “antigenoespecíficas” corren a cargo de las diversas variedades de LT y se determinan mediante la información que las CPAg (CD, MFG, etc.) proporcionan a éstos95. Cuando la oferta antigénica corresponde a epítopos bacterianos anodinos serán los LT reguladores los que intervendrán para mantener la situación de “tolerancia inmunitaria” habitual, mientras que cuando aquella oferta incluye Ag de microorganismos patógenos, serán los LT de fenotipo cooperador (CD4+), y quizá de fenotipo supresor (CD8+), los que se encargarán de montar una “reacción inmunitaria” proporcionada.
- 1)
Los LT-CD4+ son el grupo de LT numéricamente más importante en la mucosa intestinal. Esta variedad de células operativas se encuentra dispersa en la lámina propia y también salpican los folículos linfoides aislados y las placas de Peyer.
La oferta de Ag microbianos peligrosos a los LT-CD4+ vírgenes de contacto antigénico previo (células Tho) provocan su “activación” y, con esto, la secreción de IL-2 y la expresión membranaria de su receptor CD25, lo que estimula su proliferación a través de un mecanismo autocrino. De esta manera, se prepara a estos LT para iniciar su diferenciación en sus formas operativas dentro de las reacciones inmunitarias “antigenoespecíficas” dependientes de T.
Durante la década de 1990, el estudio de algunos modelos experimentales de inflamación infecciosa en ratones121 condujo al conocimiento de una dualidad evolutiva de los LT-CD4+ según las circunstancias etiológicas, que pronto se convirtió en un esquema clásico de reacción inmunológica93. Así, las infecciones provocadas por microorganismos de crecimiento intracelular originan una diferenciación de aquellas células hacia la sublínea operativa Th-1, responsable de la secreción en los tejidos inflamados de citocinas proinflamatorias, como el IFN-γ y el TNF-α, sintetizadas por estos LT-Th1 o por las células del sistema inmunitario innato (CD, MFG) que presentan los efectos de esta diferenciación. A su vez, estas últimas células apoyan la diferenciación Th-1 con citocinas inmunorreguladoras como IL-12 e IL-18, junto con el factor de transcripción T-bet123. A su vez, las lesiones granulomatosas características de estas infecciones están provocadas por aquellas citocinas proinflamatorias. Por otra parte, se encuentran las bacterias de crecimiento extracelular que son responsables de un segundo tipo de diferenciación conocido como Th-2 y, en ese caso, el tejido infectado es rico en algunas citocinas inmunoreguladoras, como la IL-4 y la IL-5 junto con la IL-13, que se comportan como antiinflamatorias y que, a través de interacciones mal conocidas, favorecen las respuestas inmunitarias humorales frente a los Ag bacterianos en juego.
En estos últimos años se ha prestado especial atención a una pequeña subpoblación de LT-CD4+ que parece protagonizar una nueva vía de diferenciación hacia una sublínea operativa, la Th-17 (así llamada por segregar la IL-17), implicada en reacciones inflamatorias, en la EII, en algunas artropatías inflamatorias y en procesos autoinmunitarios y alérgicos. Se trata de una línea celular, independiente del “doble clásico” Th-1/Th-2, altamente patogénica, que se diferencia a partir de células T-CD4+ (Tho) bajo la acción catalítica de la IL-23 y el TGF-β (transforming growth factor beta ‘factor de crecimiento transformador beta’), estas últimas, citocinas sintetizadas por CD y MFG124–126. Parece que el eje IL-23/IL-17 posee una actividad claramente inflamatoria y, a su vez, estimula la actividad fibriblástica, la de las células endoteliales, y favorece la acumulación local de granulocitos. Y todo esto lo hace bajo la acción directa de la IL-17, así como favorece la secreción por parte de otras células de la IL-1, IL-6, IL-8 y el TNF-α.
- 2)
Un segundo grupo de LT, menos numeroso y peor conocido que el anterior, es el que exhibe un fenotipo supresor (LT-CD8+); sus elementos se sitúan, preferentemente, en el interior de las células epiteliales del intestino, y su misión inmunológica es mal conocida (¿defensa frente a patógenos o lesiones locales?). Sin embargo, el hecho curioso de que en la EII se sustituyan en gran parte por LT-CD4+ indica alguna función de los primeros relacionada con la defensa de la “tolerancia inmunitaria” frente a la microflora intestinal, perdida en este proceso.
- 3)
Por último, se está empezando a estudiar un tercer grupo de LT al que se conoce como “LT reguladores”, que tiene a cargo la gestión de la “tolerancia inmunitaria” frente a Ag intraluminales (microbianos o alimentarios) anodinos93.
Dos de estos “LT reguladores” (las células Treg1 y Th-3) son de origen intestinal y atienden a Ag intraluminales debido a la secreción de 2 citocinas (la IL-10 y el TGF-β) que propician la tolerancia frente a aquellos Ag y facilitan la clonogénesis arreactiva de las células T (anergia) o la muerte celular (apoptosis) de los LT intestinales “inoportunamente” activados por Ag anodinos. Un tercer LT regulador se origina en el timo y parece gestionar la tolerancia inmunitaria a autoAg.
La situación de la “tolerancia inmunitaria” frente a los Ag de la microflora comensal fisiológica, que caracteriza la simbiosis entre esta microflora y el huésped con el que convive se rompe en la EII. De todas maneras, en la actualidad no se dispone de ninguna prueba seria de que haya una alteración funcional, de base genética o adquirida, de la subpoblación de LT reguladores. Y, sin embargo, lo que sí es evidente es que el brazo celular de la inmunidad adquirida, que corre a cargo de la población linfoide T, fundamentalmente CD4+, se comporta como “si se hubiese roto la paz y empezado la guerra” entre las 2 trincheras que separan la difícil y extensa frontera del epitelio intestinal12,127,128.
- 1)
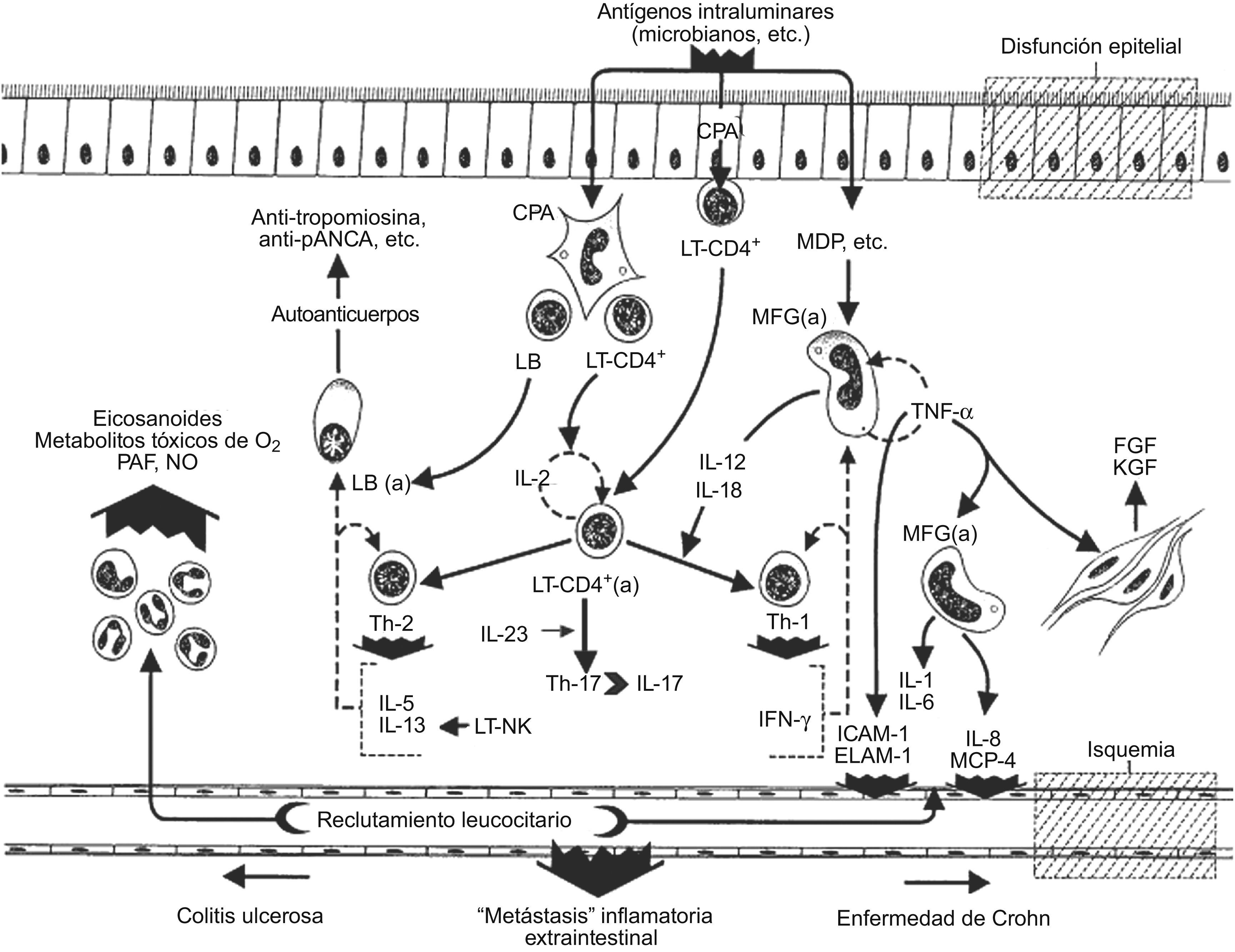
Así, los pacientes con EC exhiben un perfil de citocinas en la lesión propia de una sublínea Th-1 preferente (fig. 2), con hiperexpresión de citocinas proinflamatorias clásicas, como el IFN-γ y el TNF-α, y otras menos conocidas como las IL-21 e IL-27. Además, también se encuentra una tasa elevada de citocinas inmunorreguladoras como las IL-12 e IL-18 que, junto con el factor de transcripción T-bet (perteneciente a la familia de los factores de transcripción T-box), desempeñan un papel crucial en la diferenciación de células Th0 en Th-1. La fuerte representación de las citocinas IL-23 e IL-17 en las lesiones de este proceso hablan a favor de una participación de la vía inmunopatogénica Th-17 en la realización de la EC93,107,129–131. A su vez, los LT de la lesión mucosa de pacientes con EC son especialmente resistentes a la apoptosis debido a la hiperexpresión en éstos de la molécula antiapoptótica Bcl-2, lo que conduce a la “inmortalización” de estos linfocitos, hecho que facilita la cronificación del proceso.
- 2)
Por el contrario, en la CU, la oferta antigénica por parte de las CPAg del sistema inmunitario innato a la población de LT-CD4+ promueve una “seudodiferenciación” Th-2 algo difícil de caracterizar por ser incompleta, atípica y excepcional (fig. 2). Es una evolución Th-2 incompleta porque, aunque la mucosa colorrectal afectada es rica en IL-5 e IL-13 (citocinas Th-2 clásicas), no suele expresar un incremento de la IL-4. Es una evolución Th-2 atípica por la probable participación en la patogenia de la lesión mucosa de la CU de una pequeña fracción de linfocitos citolíticos naturales posiblemente causante de la secreción de la IL-13 con una clara actividad citotóxica sobre el epitelio colónico, hasta el punto de considerar a esta última como la citocina efectora clave de la CU. Por último, se está ante una evolución Th-2 excepcional, porque a los acontecimientos que se han comentado se suma una pincelada exótica de autoinmunidad serológica avalada por la presencia frecuente de autoanticuerpos (auto-Ac), como se verá en el apartado siguiente11,12,93,107,132.
- 3)
Todo hace pensar que a pesar de que había una idea patogénica inicial de una diferenciación Th-1 y Th-2 para la población linfoide T en la EC y la CU, respectivamente, con el tiempo se ha visto que las cosas no son tan sencillas y esquemáticas como gustaría que fuesen. Probablemente son más de 2 —y quizá mas de 4— los caminos inmunopatogénicos por donde pueden rodar los acontecimientos geneticomoleculares para “realizar” las numerosas expresiones clinicopatológicas de eso que, en un alarde de simplismo semántico, hace años se dio en llamar EII5,11,127. Y especial atención habrá que prestar al eje inmunorreactivo IL-23/IL-17; como se vio anteriormente, su papel patogénico en la EII y en otros procesos está empezando a conocerse.
La inmunidad adquirida de tipo humoral en el intestino corre a cargo de la polimorfa celularidad de LB y de su último escalón madurativo: las células plasmáticas secretoras de Ac “antigenoespecíficos”, fundamentalmente de tipo IgA.
En este apartado, se comentará la citología funcional de esta población linfoide y la participación de la inmunidad humoral en la patogenia de la EII.
Citología funcionalA diferencia de lo que ocurre con la inmunidad sistémica humoral, su representante en la mucosa intestinal exhibe una clara separación anatómica entre el “compartimento inductor” de las reacciones inmunitarias y el “compartimento efector” de éstas95.
El “compartimento inductor” está representado por los amasijos linfoides pequeños (folículos linfoides aislados) situados por millares, sobre todo en el intestino grueso, y los 200 amasijos linfoides grandes (placas de Peyer) situados en el íleon terminal133,134; unos y otros forman parte del MALT intestinal. La abigarrada población de LB se dispone en estos conexos linfoides, de dentro a fuera, como células del centro claro (centroblasto y centrocitos), células del manto y células del área marginal. Toda esta citología está recubierta de Ig de superficie, que actúan como receptoras de Ag específicos, quienes provocan una activación inmunoblástica y clonogénica de los LB llamados a responder a Ag intraluminales que han cruzado la barrera epitelial a través de las células epiteliales M que recubren los conexos linfoides.
Tras un recorrido enteroentérico más o menos extenso, los LB de este clono celular “invitado” por un Ag microbiano a dar una respuesta inmunitaria humoral “antigenoespecífica” llegan a lo que es su “compartimento efector” en los espacios subepiteliales de la lámina propia, donde se transforman en células plasmáticas. Éstas sintetizan un Ac específico, en forma de IgA dimérica, que se excreta a la luz intestinal gracias a su unión con una pieza secretora que elaboran las células epiteliales.
Participación en la patogenia de la enfermedad inflamatoria intestinalComo síntesis de la breve revisión anterior, puede decirse que, en condiciones fisiológicas, la inmunidad adquirida humoral del intestino que protagonizan los LB es de tipo IgA dimérica secretora. En cambio, en la EII, se pasa a una inmunidad sistémica de tipo IgG dominante, con cierto predominio de los Ac de tipo IgG1 e IgG3 en la CU y de tipo IgG2 en la EC. Así, en los pacientes con EII es frecuente encontrar Ac circulantes dirigidos contra Ag intraluminales microbianos. Este hecho no es más que un epifenómeno de la cadena de acontecimientos fisiopatológicos que la inflamación crónica de la mucosa intestinal origina y algunos de ellos, como los Ac anti-Saccharomyces cerevisiae, son significativamente más frecuentes en la EC que en la CU4,6,52.
Más sugerente es el desarrollo, en los pacientes afectados de CU, de un patrón de auto-Ac séricos que en algún momento ha llegado a plantear la posibilidad de que este proceso pudiese realizarse a través de un mecanismo patogénico autoinmunitario (fig. 2).
Así, en un pasado reciente, se ha encontrado un Ac sérico de tipo IgG en un 60 a un 85% de los pacientes con CU, dirigido contra una proteína de 40kD de peso molecular, aislada inicialmente en colonocitos5,6,135–137. Parece tratarse de una isoforma de la tropomiosina, concretamente de la fracción 5 de la tropomiosina humana, que es una estructura microtubular del citoesqueleto, compartida por otros colectivos celulares extraintestinales.
Por otra parte, en una proporción elevada de pacientes afectados de CU (70%) se ha podido demostrar también otro auto-Ac sérico dirigido contra la región perinuclear del citoplasma de los neutrofilos (pANCA); la probable diana de ésta es el complejo “ADN−histona H1”, componente normal de la heterocromatina del núcleo de los neutrófilos. Este auto-Ac sólo se ha encontrado en un 10% de los casos de EC5,6,138,139.
Se desconoce el significado del desarrollo en la CU —y mucho menos en la EC— de auto-Ac dirigidos frente a ciertas proteínas celulares un tanto “exóticas”, como la isoforma de la tropomiosina o una variedad de histona de la heterocromatina leucocitaria. La primera idea fue pensar en la posibilidad de que la CU fuese una enfermedad de patogenia autoinmunitaria4,140. Sin embargo, una consideración simple de los hechos indica que la CU no tiene los rasgos clásicos de las enfermedades autoinmunitarias de naturaleza humoral, como la que lucen la anemia hemolítica autoinmunitaria, la púrpura trombocitopénica idiopática o el lupus eritematoso sistémico.
El escepticismo sobre el papel patogénico de los auto-Ac en la CU ha sido apoyado desde el campo de la patología experimental93,141. Se sabe que en los ratones que carecen del gen del TCR-α−/− se desarrolla espontáneamente un cuadro de colitis, en todo idéntico al de la CU humana, incluidos sus rasgos autoinmunitarios. Cuando se cruzan ratones de esta raza con otros “mu-negativos” (mu−/−) se obtiene una cepa murina variante de la primera, que carece de células plasmáticas y, por tanto, de Ac séricos, pero que sigue desarrollando el cuadro habitual de colitis crónica. Este hecho experimental indica que, al menos en aquellos ratones, la expresión serológica autoinmunitaria no parece desempeñar un papel patogénico importante en el desarrollo de su colitis, dato que parece razonable extrapolar a la enfermedad humana.
Todo parece indicar que algunos Ag somáticos de diversa localización “mimetizan” estructuralmente a Ag de la microflora intestinal, contra los cuales, en los pacientes con CU, se han desarrollado Ac. Este hecho puede provocar una reacción serológica cruzada “seudoautoinmunitaria”4,5,93,138. De todas maneras, las Ig de estos auto-Ac, por su naturaleza IgG, a veces fijan complemento que podría originar algún daño marginal sobre los colonocitos u otras estructuras extraintestinales.
Sea como fuere, lo que sí podría ocurrir es que la “socialización” del estímulo antigénico, desde epítopos bacterianos a estructuras celulares somáticas, hiciese innecesario el papel provocador continuo de la flora intestinal para automantener una respuesta inflamatoria107.
Por último, la notable diferencia de la expresión autoinmunitaria de la CU y de la EC, a favor de la primera, debe guardar relación con la reacción inmunológica Th-2 de la segunda que, a pesar de su carácter incompleto y atípico, de alguna manera estimula las respuestas humorales características de la CU, cosa que no hace la reacción inmunológica Th-1 propia de la EC.
Riego tisular con moléculas bioactivasLa respuesta probablemente distorsionada de las células del sistema inmunitario innato (MFG y CD) y del brazo celular del sistema inmunitario adquirido (LT-CD4+) al desafío de la microflora intestinal, de algunos Ag alimentarios y a veces de ciertos auto-Ag, provoca la producción de una serie de moléculas solubles bioactivas que riegan la mucosa intestinal —y al menos en la EC, los espacios transparietales— con el impacto tisular de citocinas (fig. 2). Todo este grupo de moléculas son las que provocan, de manera inespecífica, el daño inflamatorio intestinal y no son muy diferentes de las que se encuentran como respuesta a cualquier agresión exógena. Lo excepcional en la EII es la ausencia de un claro “culpable” de la agresión y el carácter crónico y recurrente de la lesión inflamatoria142,143.
En los 2 apartados de este capítulo se resumirán brevemente los mediadores moleculares de la inflamación mejor conocidos y el papel que parecen desempeñar en la patogenia de la EC y de la CU.
Mediadores moleculares de la inflamaciónNo es fácil incluir a cada molécula conocida dentro de una familia definida, dado el papel plurivalente que algunas de ellas desempeñan y el conocimiento incompleto que todavía se tiene sobre el funcionamiento de otras. A continuación se intentará agruparlas dentro de algunas familias, por su función mejor conocida142–144.
- 1)
Citocinas proinflamatorias: IL-1, IL-6, IL-17, IL-23, IFN-γ, TNF-α, etc.
- 2)
Citocinas inmunorreguladoras : IL-2, IL-4, IL-5, IL-10, IL-12, IL-18, etc.
- 3)
Quimiocinas: péptido activador de los neutrófilos (IL-8), proteína quimioatrayente de monocitos-1 (MCP-1), etc.
- 4)
Moléculas de adherencia: ICAM-1 (intercellular adhesion molecules-1 molécula de adherencia intercelular-1'), ELAM-1 (endothelial leucocyte adhesion molecule-1 ‘molécula de adherencia endotelioleucocitaria-1’), etc.
- 5)
Factores de crecimiento: TGT-β, FGB (fibroblast growth factor ‘factor de crecimiento fibroblástico’), KGF (keratinocyte growth factor ‘factor de crecimiento queratinocítico’), PAF (platelet-activating factor ‘factor activador de las plaquetas’, etc.
- 6)
Eicosanoides: prostaglandinas (PG), tromboxanos (TX) y leucotrienos (LTN).
- 7)
Radicales libres de oxígeno y nitrógeno: anión superóxido (O−), radical hidróxilo (OH) y óxido nítrico.
El riego tisular por citocinas y otras moléculas solubles bioactivas es el escalón final de una secuencia multifásica de acontecimientos biológicos que terminan con el desarrollo de una inflamación crónica, con reactivaciones periódicas y con las imágenes de fibrosis, necrosis y angiogénesis, que caracteriza a las entidades anatomoclínicas de la EII6,107,122,127. El entramado de todas estas moléculas dibuja un sistema extraordinariamente complejo de fuerzas biológicas “enredadas” entre sí, que parecen moverse al borde del caos. Aunque hay un amplio común denominador de todas ellas en la EII, parece que el despliegue y la expresión de éstas es algo diferente en la EC y la CU (fig. 2).
Una vez más, lo que se conoce sobre esta parcela patogénica se ha aprendido del estudio cuidadoso de la patología humana de la EII y de la extrapolación a ésta de los hallazgos obtenidos a partir de algunos modelos experimentales de enterocolitis que recuerdan la EC y la CU humanas6,8,10,107,122.
- 1)
Así, tal como se vio en el capítulo anterior, en la EC domina la expresión tisular de ciertas citocinas, como las IL-12, IL-18, IFN-γ, TFN-α, IL-23 e IL-17. Por el contrario, en la CU suele haber una expresión dominante de la IL-5 e IL-13, mientras que la IL-4 sólo está elevada en casos aislados sin clara relación con el grado de inflamación6,93,107,142–144.
- 2)
Las IL-1 e IL-6 se expresan por igual en ambos procesos, y se comportan como reactantes inespecíficos de fase aguda. Lo mismo ocurre con la IL-2, como primera expresión de la activación de los LT-CD4+, y la IL-10, ejemplo de citocina antiinflamatoria expresada por igual en cualquier forma de EII.
- 3)
La expresión por parte de células del sistema innato (MFG, etc.) de moléculas de adherencia, como ICAM-1 y ELAM-1, y de moléculas citoatrayentes, como la IL-8 y el MCP-1, facilitan el contacto leucocitario con el endotelio vascular y el reclutamiento de fagocitos sanguíneos (neutrófilos y monocitos) hacia los territorios enfermos de la pared intestinal. No parece haber una clara diferencia en la expresión tisular de estas moléculas entre la CU y la EC; a pesar de ésta, la infiltración de polimorfonucleares parece ser más frecuente y llamativa en los brotes de la CU que en los brotes de la EC122,142,144,145.
- 4)
A partir de las células de este infiltrado de fagocitos profesionales, y muy fundamentalmente de polimorfonucleares, se generan lo que podría llamarse los últimos mediadores de la lesión tisular, y también, como se verá después, algunos mediadores de la reparación de la pared intestinal107,122,144,146. Entre ellos, destaca la familia de moléculas conocidas como eicosanoides (es decir, las PG, los TX) y leucotrienos (LTN), productos terminales de la degradación de los fosfolípidos membranarios de los leucocitos “gastados” en la inflamación, después de transformarse en ácido araquidónico metabolizado por la vía de la ciclooxigenasa (PG y TX) y de la lipooxigenasa (LTN). La mayoría de estos productos terminales tienen una actividad proinflamatoria, aunque algunos, como la PG-E2, parecen comportarse como antiinflamatorios.
Los eicosanoides, al margen del signo de su actividad, se encuentran uniformemente elevados en la EII, aunque se apuntan algunas pequeñas diferencias. Así, la PGE2, el TX-B2 y el LTN-B4 están más elevados en la CU que en la EC. Algo parecido ocurre con el PAF, otro mediador inespecífico de la inflamación intestinal: su expresión parece ser mayor en la CU.
- 5)
Se está prestando una atención creciente a la contribución de los leucocitos polimorfonucleares en la patogenia de la EII, desde el momento en que se sabe que son ellos la fuente principal de una serie de moléculas citotóxicas derivadas del oxígeno (O2), como el O− y el OH, con capacidad lesiva sobre la pared intestinal107,122,142. El eje inmunorreactivo IL-23/IL-17 desempeña un papel muy importante en esta “atracción leucocitaria”.
Hay pruebas procedentes de modelos experimentales de que estos radicales tóxicos del O2 están involucrados en las enterocolitis de ratones, similares a la EII humana, e igualmente se han encontrado, con técnicas de quimioluminiscencia, grandes concentraciones de éstos en la mucosa de pacientes con EC y CU, en relación directa con la gravedad de la enfermedad. Es posible que la falta de antioxidantes en la mucosa de estos pacientes la haga más susceptible a cualquier lesión oxidativa.
- 6)
Hay también ciertas pruebas, procedentes de modelos experimentales, de que el óxido nítrico (que se comporta como una molécula bioactiva y de señales en el tubo digestivo) está involucrada en la inflamación que caracteriza a las enterocolitis murinas. Apoya esta idea el hecho de que la inhibición de la “sintasa del óxido nítrico inductible” con análogos de la arginina puede aminorar la gravedad de aquella inflamación10,22,142,146.
A pesar de todo, no está claro el papel del óxido nítrico en la EII; incluso se ha especulado sobre un posible “efecto dual”, a la vez tóxico y protector. Por una parte, la probable depleción de antioxidantes de la mucosa de estos pacientes la haría más susceptible del efecto nocivo del óxido nítrico y, por otra, este metabolito ejercería un efecto protector de la inflamación intestinal, lo que inhibiría el reclutamiento leucocitario y actuaría como quelante de radicales libres de O2.
- 7)
Por último, hay una serie de péptidos que parecen trabajar no como agentes lesivos, sino como mediadores de la reparación tisular147,148. Esto se refiere, en primer lugar, a una serie de factores de crecimiento, como el TGF-β, el FGF y el KGF, junto con otras moléculas como el “péptido trifoliado”, la IL-10, la PG-E2 y el mismo óxido nítrico, del que se habló antes.
Quedaría incompleto el argumento de este trabajo si no se comentara, aunque fuese brevemente, el posible papel de la isquemia en la patogenia de la EC y la significación biológica de las manifestaciones clinicopatológicas extraintestinales en la EII.
La isquemia como parcela patogénicaSe ha especulado sobre la posibilidad de que un factor de isquemia parietointestinal y mesentérica pudiese desempeñarse en el desarrollo de la EC, lo que afectaría de manera difusa a pequeños vasos72,149–151. Entre un 3 y un 85% de las piezas de resección quirúrgica de pacientes con EC exhiben lesiones inflamatorias de los vasos sanguíneos que se localizan, de forma característica, en la cara externa de la pared intestinal. A su vez, en un 3 a un 20% de estos casos se han podido observar lesiones granulomatosas de las arterias y venas mesentéricas y de la serosa. Todo esto acompañado, en ocasiones, de imágenes microobstructivas. Por supuesto, estas lesiones no suelen encontrarse en la CU.
De acuerdo con este modelo patogénico se pensó que podría actuar el virus del sarampión a través de una infección pesistente (vasculitis granulomatosa vírica). También se ha sospechado que el efecto inductor del tabaquismo y la toma de anticonceptivos orales en la realización de la EC podrían tener su explicación en lesiones de microtrombosis múltiple, a través de un estado de hipercoagulabilidad.
Sin poder negar la realidad de esta posible parcela patogénica de la EC, es presumible que se trate de un acontecimiento marginal que puede desempeñar algún papel en un número limitado de casos, sometidos a circunstancias etiológicas especiales.
Metástasis inflamatoria extraintestinalEs relativamente frecuente (entre un 20 y un 35% de los casos) la presentación de manifestaciones clínicas extraintestinales en pacientes afectados de EII, tanto en casos de CU como de EC, aunque con algunos matices diferenciales entre ambos procesos152,153. Este acontecimiento puede afectar a cualquier territorio orgánico de la economía humana, aunque sus localizaciones y formas preferentes se presentan en la piel (eritema nodoso y pioderma gangrenosa), las estructuras osteoarticulares (artritis, osteopatías hipertróficas), los ojos (uveítis y epiescleritis) y el hígado (colangitis esclerosante y cirrosis biliar primaria). Estas “metástasis inflamatorias” dan una proyección curiosamente sistémica a una enfermedad primitivamente intestinal.
Es difícil saber cuál es el significado biológico de estas manifestaciones extraintestinales. Para algunos autores, se estaría ante una activación sistémica tanto de células inmunomoduladoras como de cascadas enzimáticas (complemento, kalicreína, etc.), provocada por la entrada masiva de moléculas antigénicas a través de una barrera intestinal enferma y permeable. Para otros autores, este hecho sería una consecuencia más de la “socialización antigénica”, al compartir diversos Ag somáticos una estructura molecular parecida a la de los Ag microbianos que provocaron el conflicto inmunológico original. La reacción inmunitaria (celular y humoral) frente a estos últimos afectaría, por un mecanismo de “mirón inocente”, a los primeros.
Horizonte de una historia inacabada- 1)
Todo parece apuntar —por lo que se ha ido viendo en los diversos capítulos de esta revisión— que la EII traduce la expresión de un conflicto surgido entre 2 “trincheras biológicas” que normalmente conviven en paz: de un lado, la microflora intestinal (junto con otros Ag intraluminales) y de otro lado, el sistema inmunitario local del huésped. Todo esto, modulado por numerosos factores ambientales y al amparo de ciertos factores poligénicos de susceptibilidad1,3,5,12,107,143.
En esencia, este conflicto podría surgir por una disfunción del sistema inmunitario innato o adquirido (celular o humoral) frente a bacterias comensales normales, por una respuesta inmunitaria apropiada frente a una microflora bacteriana que ha variado en su composición absoluta o relativa, por una barrera epitelial funcionalmente alterada o por todo a la vez, en una secuencia multifásica difícil de hilvanar154. La “socialización” de la provocación antigénica desde epítopos microbianos iniciales a epítopos enterocolónicos y extraintestinales, después explicaría la cronicidad del proceso y su aproximación, en bastantes casos, al perfil de una enfermedad sistémica. Este parece ser “el hoy” de la EII.
- 2)
Con toda seguridad, ésta no es toda la verdad y quizá sólo sea una parte de ésta. Es evidente que las circunstancias etiológicas desde las que se puede realizar la EII y las parcelas patogénicas donde se realiza son múltiples y no se excluyen entre sí. Y no sería de extrañar que en un futuro no muy lejano se llegase a la conclusión de que no se está ante una enfermedad en singular (la EII) ni tampoco ante la simple dicotomía de una pareja de enfermedades “primas hermanas” (la EC y la CU), sino ante un amplio espectro de procesos patológicos que surgen desde factores etiológicos y caminos patogénicos diferentes, para terminar en un hecho biológico con un común denominador que es la inflamación crónica y recurrente de la pared intestinal con posibles “metástasis inflamatorias” a distancia3,5,9,107. Algunos ejemplos de esta diversidad empiezan a dibujarse en el horizonte clínico de lo que puede ser “el mañana” de la EII.
- 3)
Por último —y al considerar el argumento de este trabajo desde una perspectiva más distante—quizá, en el fondo, lo que ocurre es que se asiste, sin que uno se dé cuenta, a las consecuencias de un “cambio de signo” de la morbilidad de los seres humanos experimentado durante el último siglo. Esto hace referencia a que el descenso o incluso la desaparición de numerosas enfermedades infecciosas en los países desarrollados podría estar siendo ocupada por un incremento de las enfermedades inflamatorias, alérgicas y autoinmunitarias, entre sus habitantes (EII, asma, diabetes de tipo 1, esclerosis múltiple, etc.).
Parece como si la mejor calidad del agua y la higiene en general, el descubrimiento de los antibióticos y la mejoría de las pautas de asistencia sanitaria en la infancia de los habitantes del mundo desarrollado, causantes del dramático descenso de las enfermedades infecciosas entre sus habitantes, hubiesen dejado sin su trabajo ancestral al sistema inmunitario de éstos y éste se hubiese visto obligado, como “alternativa laboral”, a montar reacciones inmunológicas dislocadas frente a microorganismos comensales del intestino, a Ag alimentarios y ambientales o incluso frente a auto-Ag inocentes. Éste es —medio en broma, medio en serio—el fondo de la llamada “teoría de la higiene”, hipótesis provocativa, donde las haya, y difícil de probar 95,155–157, pero que invita a meditar sobre el precio que a veces hay que pagar por los avances médicos de los que con frecuencia uno se siente orgulloso. Éste podría ser el “pasado mañana” de la EII.

