




El diagnóstico de la enfermedad celíaca resistente (ECR) se establece, tras la exclusión de otras entidades, ante la persistencia de datos clínicos de malabsorción y atrofia vellositaria durante 6-12meses a pesar de una estricta dieta sin gluten (DSG). La detección de alteraciones en la población linfocitaria intraepitelial es importante para su diagnóstico. Un subgrupo de pacientes con ECR pueden desarrollar complicaciones severas, como linfoma T asociado a enteropatía (LTAE). Presentamos el caso de un paciente con EC silente de larga evolución que finalmente derivó en LTAE y que evidencia el reto que supone para el clínico tanto el diagnóstico como el tratamiento de esta entidad.
Diagnosis of refractory celiac disease (CD) is based on exclusion of other disorders, persistence of malabsorptive symptoms and villous atrophy, despite a strict gluten-free diet for at least 6-12months. Detection of alterations in the intraepithelial lymphocyte population is crucial for diagnosis. A subgroup of patients with refractory CD may develop severe complications such as enteropathy-associated T cell lymphoma (EATL). We present the case of a patient with longstanding silent CD who developed EALT, highlighting the challenge posed by the diagnosis and treatment of this entity.
La enfermedad celíaca (EC) es un trastorno de naturaleza autoinmunitaria desencadenado por la ingesta de gluten en individuos genéticamente predispuestos. La enfermedad celíaca resistente (ECR) se define como aquella EC que no mejora clínica ni histológicamente tras al menos 6-12meses de dieta exenta de gluten (excluyendo la ingesta de gluten de forma inadvertida, otras causas de atrofia vellositaria y el linfoma intestinal manifiesto). La ECR se subdivide en ECR-I, caracterizada por una población normal de linfocitos intraepiteliales (LIE), y ECR-II, que suele presentar LIE inmunofenotípicamente aberrantes y de carácter monoclonal. La ECR-II evoluciona con mayor frecuencia hacia el LTAE, lo que ensombrece su pronóstico. El objetivo de este trabajo es presentar un caso de ECR asociada a LTAE que presentó especial dificultad diagnóstica y planteó la cuestión de cuándo y cómo debe iniciarse el tratamiento antineoplásico de esta entidad.
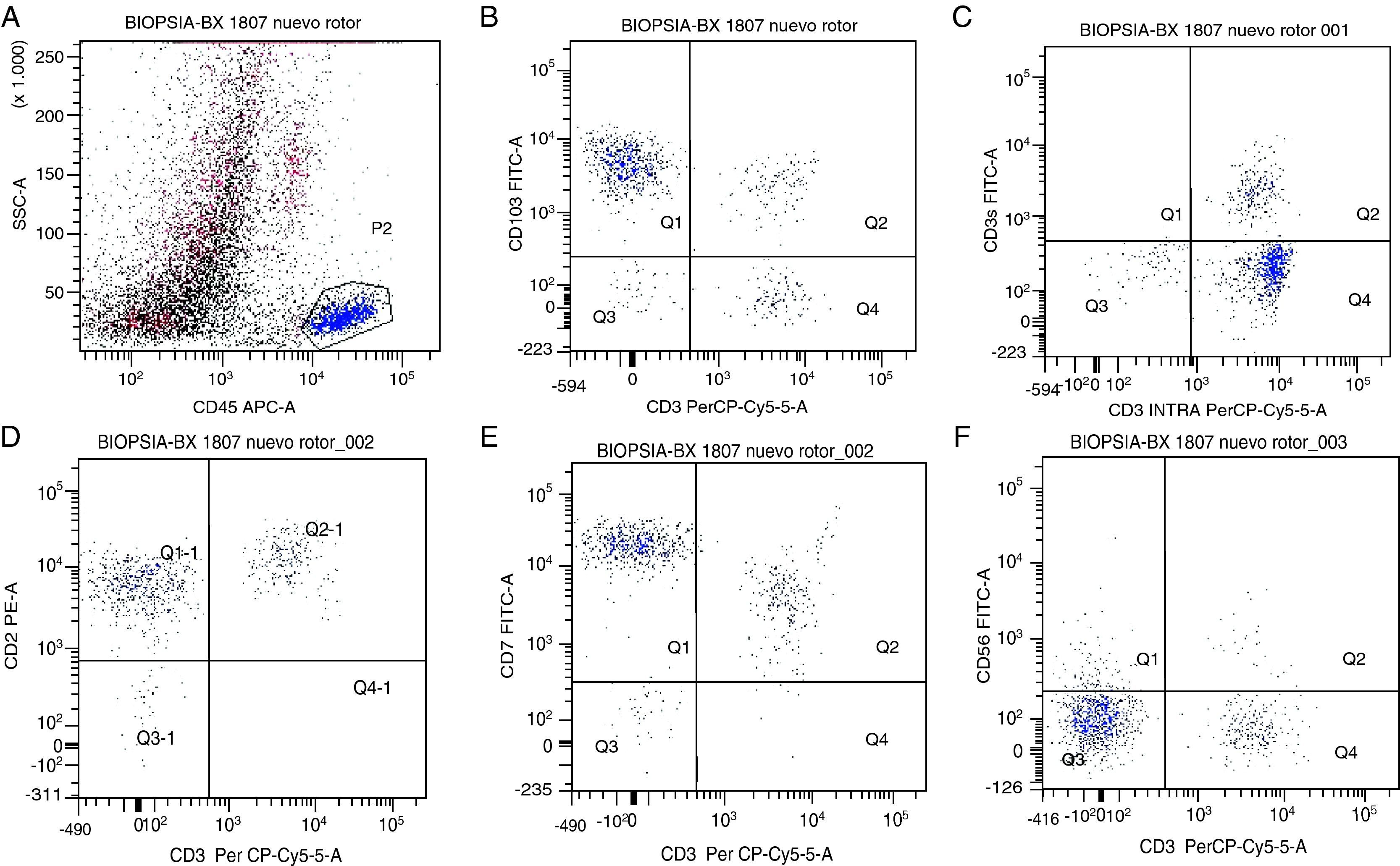
Observación clínicaVarón de 46 años sin antecedentes personales ni familiares de interés que acude a consulta por epigastralgia y vómitos posprandiales de un año de evolución. No refería cambios en el ritmo intestinal ni síndrome constitucional asociado y nunca antes había presentado sintomatología digestiva alguna. No presentaba fiebre ni alteraciones cutáneas. En la exploración se evidenciaron signos de desnutrición marcada y adenopatías palpables en las regiones retroauricular, laterocervical y axilar. El abdomen estaba levemente distendido, con ruidos hidroaéreos aumentados pero sin masas ni megalias palpables. El resto de la exploración física no mostró alteraciones relevantes. Se realizó una analítica completa, que corroboró el estado de desnutrición del paciente: proteínas totales 6g/dl, albúmina 2,2g/dl, proteína ligada al retinol 1,2mg/dl, prealbúmina 9,5mg/dl, colesterol total 105mg/dl, triglicéridos 42mg/dl y ácido fólico 1ng/ml. Los parámetros de función hepática y renal, los electrolitos, el perfil tiroideo y el estudio de coagulación fueron normales. Además, se objetivó un aumento de IgG (2.420mg/dl) e IgA (495mg/dl), sin banda monoclonal significativa en la inmunofijación del suero. El hemograma mostró hemoglobina, VCM y CHCM normales, junto con ligera leucocitosis (12.700 cél./μl, con linfocitosis verdadera de 5.300cél./μl) y trombocitosis (583.000cél./μl). La PCR y la VSG se encontraban dentro del rango normal (2,45mg/l y 10mm, respectivamente). La cifra de grasa en heces estaba dentro de la normalidad (5g/24 h). Se solicitaron anticuerpos antiendomisio y antitransglutaminasa IgA que resultaron positivos y se hizo un estudio de los genes HLA asociados con la enfermedad celíaca, hallando que el paciente era doble DQ2. La panendoscopia oral evidenció escasez de pliegues intestinales. Las biopsias de la segunda porción mostraron atrofia de vellosidades, hipertrofia de criptas y aumento de LIE, hallazgos compatibles con EC grado 3C de la clasificación de Marsh. El estudio inmunofenotípico por citometría de flujo de los LIE de estas primeras biopsias (fig. 1) evidenció en ese momento la existencia de una población linfoide aberrante CD3−, CD103+, CD56− y CD3¿+, que constituía un 69% de los LIE. Además, se detectó un reordenamiento clonal de la cadena gamma del TCR. Se estableció el diagnóstico de EC y el paciente comenzó una DSG, con buena adherencia. Dos meses más tarde su estado general había mejorado, la epigastralgia había desaparecido y había recuperado 3kg de peso. Es de destacar que su EC, si bien no podía calificarse aún de resistente, pues todavía no habían transcurrido 6 meses de tratamiento con DSG, mostraba ya a nivel molecular datos de riesgo para evolucionar de forma complicada (una población linfoide aberrante), a pesar de lo cual no se modificó la actitud terapéutica, dada la ausencia de recomendaciones consensuadas sobre las pautas a seguir ante este hallazgo.
 que refleja los LIE seleccionados para el análisis en función de su ángulo de dispersión lateral de luz y a la expresión del marcador panleucocitario CD45. -Paneles B y C: representan la presencia de un elevado porcentaje de una subpoblación LIE CD3− CD103+ y la expresión intracitoplasmática de CD3( en casi la totalidad de las células CD3− en superficie. -Paneles D, E y F: muestran la expresión positiva de CD2 y CD7, y negativa para CD56 de la subpoblación LIE CD3− CD103+ analizada.")
Estudio inmunofenotípico de los LIE por citometría de flujo.
-Panel A: gráfico de puntos (dot-plot) que refleja los LIE seleccionados para el análisis en función de su ángulo de dispersión lateral de luz y a la expresión del marcador panleucocitario CD45.
-Paneles B y C: representan la presencia de un elevado porcentaje de una subpoblación LIE CD3− CD103+ y la expresión intracitoplasmática de CD3( en casi la totalidad de las células CD3− en superficie.
-Paneles D, E y F: muestran la expresión positiva de CD2 y CD7, y negativa para CD56 de la subpoblación LIE CD3− CD103+ analizada.
Seis meses más tarde el paciente requirió ingreso por nuevo deterioro de su estado nutricional, fiebre ocasional y persistencia del dolor abdominal. Desde el diagnóstico había seguido la DSG de manera estricta y los anticuerpos antitranglutaminasa IgA se habían negativizado. Durante su hospitalización continuó con DSG y se pautaron suplementos dietéticos, pero continuó perdiendo peso y comenzó a presentar diariamente picos febriles nocturnos. Se realizó una TC toraco-abdomino-pélvica que mostró adenopatías de gran tamaño inguinales y en la raíz del mesenterio. Se efectuó un tránsito intestinal, que evidenció engrosamiento de pliegues y dilatación de asas de intestino delgado. Se volvieron a obtener biopsias duodenales, sin cambios ni en el grado de atrofia ni en las características de la población linfoide con respecto a las previas. Se llevó a cabo una valoración del intestino delgado mediante cápsula endoscópica que identificó atrofia vellositaria desde duodeno hasta tramos muy distales, sin irregularidad de pliegues, úlceras o nódulos que indicasen degeneración. El estudio de leucocitos de sangre periférica encontró un ligero aumento de linfocitos T activados, pero sin alteración en sus antígenos o en el porcentaje de las diferentes poblaciones. El aspirado medular no reveló hallazgos patológicos. Los estudios microbiológicos (micobacterias, malaria, Leishmania spp., Salmonella spp., Brucella spp., fiebre Q y Aspergillus fumigatus) resultaron negativos. A las 5 semanas de ingreso, el paciente presentó una lesión cutánea en el flanco izquierdo de 2cm de diámetro, sobreelevada, indurada, descamada y de color violáceo, cuya biopsia fue compatible con linfoma. Los linfocitos de esta lesión presentaban el mismo inmunofenotipo y el mismo reordenamiento de la cadena gamma del TCR que los linfocitos de las biopsias intestinales. De este modo, el paciente fue diagnosticado de LTAE, iniciándose quimioterapia tipo CHOP (ciclofosfamida, doxorrubicina, vincristina y prednisona). Un año después falleció debido a una infección grave por Clostridium difficile, asociada a bronconeumonía y a infarto esplénico masivo.
DiscusiónLa EC puede presentar distintas complicaciones entre las que destaca el LTAE, entidad rara en la población general (incidencia de 0,1/100.000habitantes en Holanda) pero cuya frecuencia en la EC globalmente considerada es de alrededor del 5%1. Como otras complicaciones asociadas a la EC el LTAE es más frecuente en la ECR2. La ECR-I tiene un riesgo de linfoma mayor que la forma clásica, pero mucho menor que la ECR-II, entidad que hoy es considerada como un linfoma T críptico, puesto que en los individuos que la presentan el LTAE puede ocurrir hasta en un 60-80% de los casos a los 5 años3–6.
La presentación clínica del LTAE suele ser insidiosa y con frecuencia se manifiesta como una recidiva clínica (diarrea, dolor abdominal, pérdida de peso, etc.) en pacientes celíacos en los que la dieta sin gluten había controlado los síntomas hasta ese momento. En otras ocasiones, se manifiesta con fiebre, linfadenopatía mesentérica con cavitación central, obstrucción intestinal o hemorragia digestiva4,5. El pronóstico de los pacientes con LTAE es pobre, con supervivencia global aproximada a 2años del 28%3,6,7. La quimioterapia sistémica es una opción terapéutica para estos pacientes, dado que suelen ser tumores multifocales y presentar extensión ganglionar en el momento del diagnóstico.
Actualmente parece claro que el origen del linfoma se encuentra en los LIE8–11. El proceso se iniciaría a partir de una población aberrante de LIE, con inmunofenotipo y genotipo diferentes a los de los LIE de individuos sanos, enfermedad celíaca clásica o ECR-I, pero muy similares a las de los LIE de la ECR-II. En concreto, esas características anómalas se expresan como pérdida del CD3, CD8 y TCR de la superficie celular (conservando la expresión del CD3 citoplasmático y el CD7 de membrana), y el reordenamiento monoclonal de los genes del TCR. Los linfocitos aberrantes, desde momentos previos a la aparición del linfoma, se encuentran ya entre los LIE de amplias áreas de la mucosa gastrointestinal, no sólo allí donde surgirá el linfoma, siendo su detección indicativa de un mayor riesgo de que la EC acabe complicándose con un LTAE12. Así, por ejemplo, el trabajo de Verbeek et al.13 muestra que la detección de más de un 20% de LIE CD3−, CD3¿+ y CD7+ posee gran sensibilidad y valor predictivo negativo para predecir el desarrollo de un LTAE. Del mismo modo, del estudio de Liu et al.14 se desprende que la presencia de más de un 80% de LIE con pérdida del CD8 de membrana y expresión del CD3 citoplasmático también predice fuertemente la evolución hacia LTAE. Esto supone una ayuda de cara a la detección de un linfoma eventual, pues aunque la identificación de estos linfocitos aberrantes en las biopsias de la mucosa intestinal no sea diagnóstica del mismo, sí constituye un marcador de riesgo14–17.
Centrándonos en nuestro caso clínico, nos gustaría resaltar que se trataba de un paciente con EC que durante años se mantuvo silente. Por este motivo no consultó, retrasándose el diagnóstico y el inicio de la DSG, hecho que muy probablemente contribuyó a que la evolución fuese tórpida, con evolución hacia un LTAE. En efecto, uno de los factores más fuertemente asociados al desarrollo de linfoma intestinal en los pacientes con EC es la ausencia de una DSG. La ingesta continuada de gluten constituye un estímulo antigénico constante para los linfocitos de la mucosa entérica, contribuyendo a que éstos pierdan los mecanismos que controlan su proliferación y se tornen anómalos, hasta poder dar lugar a un linfoma. De todos modos, es conocido que un gran número de pacientes celíacos permanecen sin diagnóstico y sin DSG durante largos periodos de tiempo y nunca desarrollan un LTAE. Desconocemos qué factores, además de persistencia en la ingesta de gluten, contribuyen al desarrollo final de complicaciones en la EC.
Por otro lado, no podemos dejar de mencionar el hecho de que el principal obstáculo que plantea el LTAE es el problema de su confirmación diagnóstica. No obstante, aunque todavía no se ha identificado un patrón de alteraciones en los linfocitos de la mucosa gastrointestinal que indiquen de forma inequívoca la presencia de un linfoma en el tracto digestivo, numerosos estudios muestran que un buen porcentaje de los pacientes cuyas biopsias presentan linfocitos aberrantes acaban desarrollando LTAE. Es decir, comenzamos a disponer de una herramienta que puede facilitar el diagnóstico precoz de esta entidad, que es el análisis de los linfocitos de la mucosa entérica. Sin embargo, no existe un consenso claro acerca de cuál debe ser la actitud a tomar ante la presencia de linfocitos aberrantes, especialmente acerca de si este hallazgo debe dar lugar al inicio de medidas terapéuticas18–20. De hecho, en nuestro caso se detectaron linfocitos aberrantes ya desde la primera biopsia, pero ello no dio lugar a cambios en nuestro proceder, precisamente por la ausencia de recomendaciones en esta materia. Por ello creemos que la significación de la presencia de linfocitos aberrantes en la mucosa gastrointestinal debería ser revisada y que convendría establecer unas directrices acerca de las implicaciones prácticas de este hallazgo.

